Genome-Wide Identification and Expression Analysis of Aspartic proteases in Populus euphratica Reveals Candidates Involved in Salt Tolerance
Peiyang He, Lifan Huang, Hanyang Cai

TL;DR
This study identifies and analyzes aspartic protease genes in Populus euphratica, revealing their potential role in salt tolerance and offering candidates for improving woody crops.
Contribution
The first comprehensive analysis of aspartic protease genes in Populus euphratica, linking them to salt tolerance and genetic engineering potential.
Findings
55 PeAP genes were identified and classified into three classes based on conserved structures.
PeAP genes show conserved evolution and variable regions, suggesting functional diversity.
PeAPs are linked to cis-elements that may mediate responses to stress and phytohormones.
Abstract
Aspartic proteases (APs) are among the four primary families of proteolytic enzymes found in plants, and they are essential for both stress response mechanisms and developmental activities. While the AP gene family has been studied in model plants like Arabidopsis, its characterization in woody species-particularly in extremophytes like Populus euphratica, remains limited. Moreover, the potential involvement of APs in salt tolerance mechanisms in trees is yet to be explored. In this research, 55 PeAPs were discovered and categorized into three distinct classes based on their conserved protein structures. The phylogenetic analysis revealed potential functions of AP genes derived from Arabidopsis thaliana, V. vinifera, and P. euphratica. Our findings indicate that PeAP possesses a well-conserved evolutionary background and contains numerous highly variable regions, making it an excellent…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
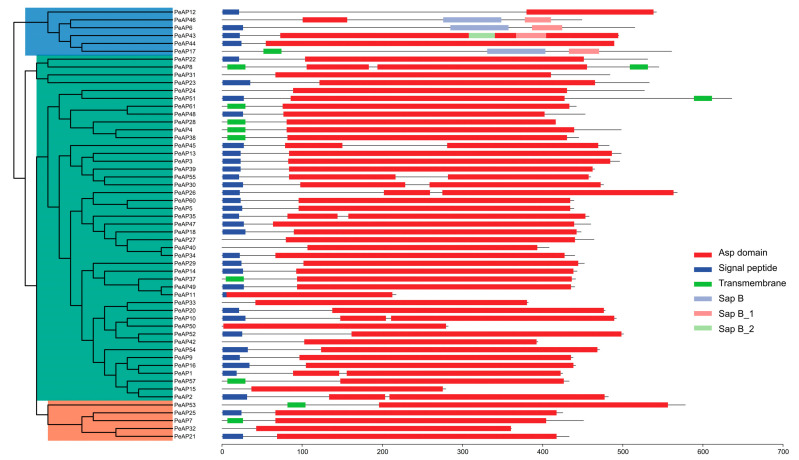 Figure 1
Figure 1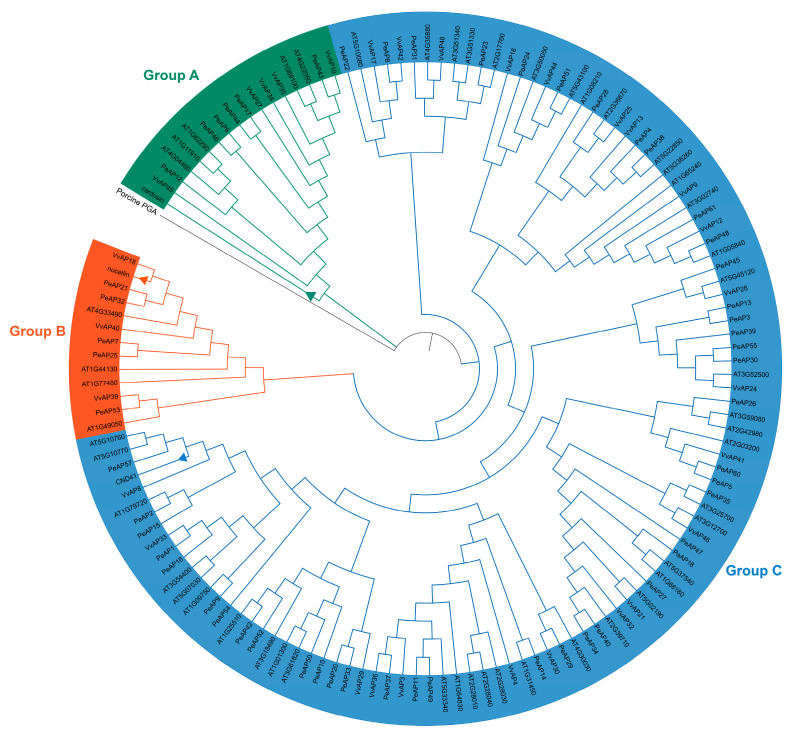 Figure 2
Figure 2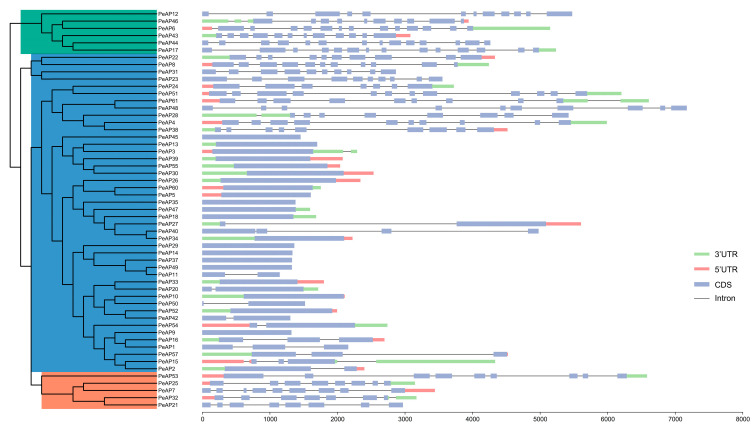 Figure 3
Figure 3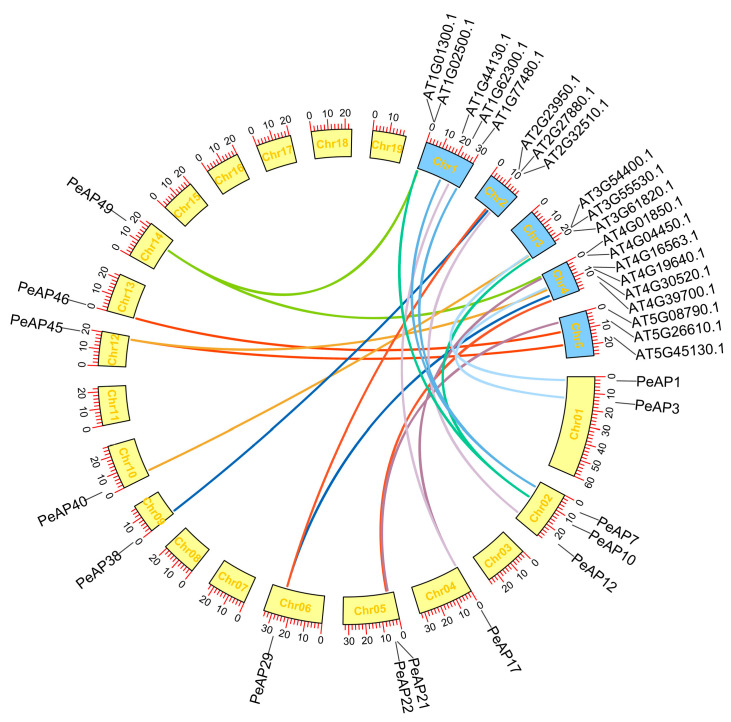 Figure 4
Figure 4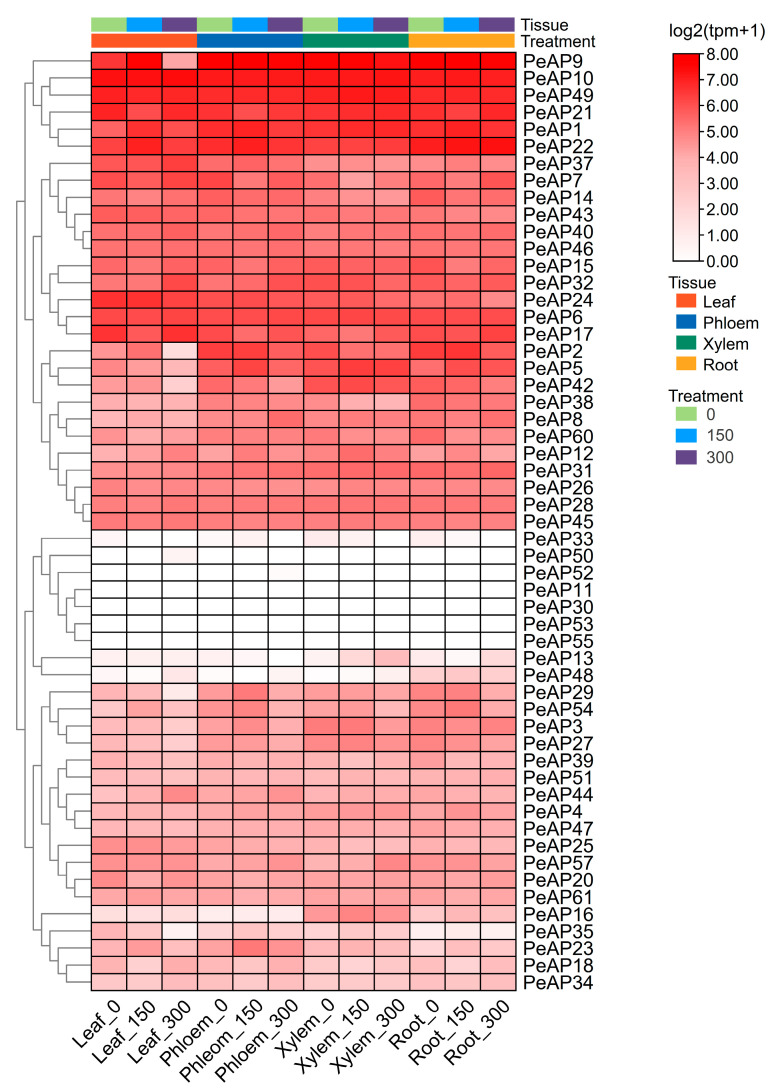 Figure 5
Figure 5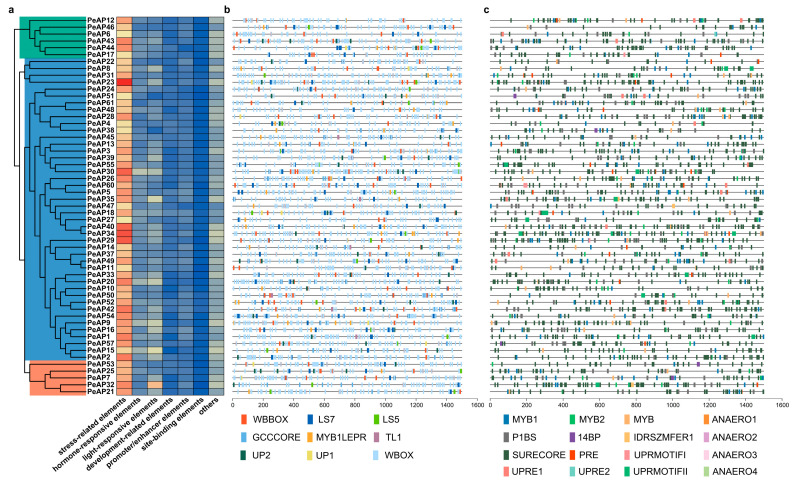 Figure 6
Figure 6- —Major Science and Technology Project of Fujian Province
- —National Natural Science Foundation of China
- —Fujian ‘Young Eagle Program’ Youth Top Talent Program
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPhotosynthetic Processes and Mechanisms · Phytase and its Applications · Plant Reproductive Biology
1. Introduction
Soil salinity has emerged as one of the most critical abiotic stresses threatening global forest ecosystems, with recent estimates suggesting that over 20% of irrigated agricultural lands worldwide are affected by secondary salinization [1]. This environmental challenge is particularly acute for woody perennials, where prolonged exposure to saline conditions disrupts physiological processes ranging from nutrient acquisition to vascular development [2,3,4]. Among tree species, Populus euphratica has garnered significant scientific attention due to its remarkable capacity to thrive in saline-alkali soils with NaCl concentrations exceeding 400 mM—a trait attributed to its sophisticated ion homeostasis mechanisms [5]. Recent studies have elucidated several components of this halophytic adaptation system, including the SOS pathway-mediated Na^+^ exclusion, enhanced antioxidant enzyme activities, and developmental plasticity in root architecture [4]. However, despite these advances, the proteolytic regulation underlying P. euphratica’s salt tolerance remains largely unexplored, particularly concerning the aspartic protease (AP) gene family, which has been associated with stress responses across various plants [6,7].
Aspartic proteases represent one of four major classes of proteolytic enzymes in plants, characterized by their conserved biological structure containing two catalytic aspartate residues in the characteristic DTG/DSG motifs [8,9]. Phylogenetic analyses reveal that this gene family has undergone significant expansion during land plant evolution, with modern species displaying remarkable diversity in AP gene copy numbers—from 51 members in Arabidopsis to 96 in Oryza sativa [10,11]. Beyond their canonical roles in protein turnover and processing, plant APs have been increasingly recognized as key regulators of stress responses through various mechanisms. In Arabidopsis thaliana ectopic expression of a grape aspartic protease gene, AP13, improves resistance to powdery mildew by promotes the SA dependent signal transduction pathway, but suppresses the JA signal transduction pathway [12]. In addition, ASPG1 (NP_188478) the ASPARTIC PROTEASE IN GUARD CELL 1 gene whose over expression conferred drought avoidance via ABA-dependent signaling in Arabidopsis [13]. Perhaps most intriguingly, certain AP isoforms in Arabidopsis (AtPCS1) and sweet potato (SpAP1) have been demonstrated to influence developmental processes ranging from embryogenesis to leaf senescence [14,15], suggesting evolutionary co-option of proteolytic functions for regulatory purposes.
In the context of P. euphratica’s salt tolerance, several molecular adaptations have been characterized that may intersect with AP functionality. The species exhibits sophisticated transcriptional reprogramming during salt stress, involving NAC and WRKY transcription factors that regulate ion transporter genes such as NHX1 and HKT1 [16,17]. Post-translational modifications (PTMs) also play crucial roles, with recent studies identifying stress-responsive deubiquitination of some proteins [18] and glycosylation of membrane proteins [19]. Furthermore, while the Populus trichocarpa genome project revealed extensive gene family expansion through segmental duplications [20], the evolutionary trajectory and functional specialization of AP genes in its extremophyte relative P. euphratica have never been systematically investigated.
The current understanding of plant APs presents several critical knowledge gaps that this study addresses. First, while AP family members have been comprehensively annotated in model herbaceous species like Arabidopsis [10] and rice [11], their identification and classification in extremophyte trees like P. euphratica remains incomplete, hindering comparative evolutionary analyses across ecological niches. Second, the potential involvement of APs in woody plant stress responses has been largely overlooked, despite emerging evidence of their regulatory roles in herbaceous species [21]. Third, the relationship between AP gene family expansion through whole genome duplication events [20] and functional diversification in stress adaptation contexts remains unclear. Finally, while post-translational modifications like glycosylation are known to regulate AP activity in some microbials [22], no studies have explored the intriguing possibility that AP function might be modulated by glycosylation, which was a key salinity-responsive PTM in P. euphratica. These gaps collectively represent a significant limitation in our understanding of plant stress biology, particularly for ecologically and economically important tree species facing increasing salinity pressures [23], where APs may serve as underexplored nodes in stress signaling networks.
In this study, we conducted a comprehensive analysis of the complete genome of P. euphratica, focusing on the identification and characterization of the PeAP gene family. Our investigation delved into the phylogenetic relationships among these genes, as well as their structural properties at the protein level. We also explored transcriptional patterns, the presence of cis-acting elements within their promoters, and the subcellular localization of the proteins were encoded by these genes. Additionally, we examined potential interactions that may occur among these proteins. Through this extensive analysis, we identified a specific group of PeAP genes that appeared to play significant roles in the ability of medicinal plants to adapt and respond to varying environmental conditions.
2. Results
2.1. Genome-Wide Identification and Phylogenetic Analysis of P. euphratica AP Gene Family
In order to pinpoint members of the AP family within the genus Populus, a comprehensive BLASTP analysis was conducted. This analysis utilized previously documented AP protein sequences from Arabidopsis as the query for comparison. We discovered and characterized a total of 61 APs in P. euphratica, using APs from Arabidopsis thaliana and Vitis vinifera as references (Table S1). The findings of this analysis revealed that a significant number of the identified proteins, specifically, a total of 55 PeAPs, exhibited high sequence conservation, indicating evolutionary preservation. This conservation aligns with the APs that have been previously identified in A. taliana. Based on these characteristics, the 55 PeAPs, as well as other known AP proteins (Nucellin, CND41 and cardosin A) were categorized into three distinct groups, labeled A, B, and C, similar to those described in Arabidopsis and grape. Every group had individual conserved domains, which reinforced the relevance of the classification (Figure 1). Approximately 94.5% of the PeAP CDSs encode more than 360 aa long, while PeAP11 (217 aa), PeAP15 (279 aa), and PeAP50 (282 aa) were unique (Table S1). The protein sequences within each category exhibited significant similarities. The PeAPs molecular weights ranged between 23.4 and 70.8 kDa. Isoelectric points showed variation from 4.65 to 9.78. Among them, PeAP51 encodes the heaviest protein at 70.8 kDa, while PeAP11 encodes the lightest at 23.4 kDa (Table S1). The characteristics of the PeAPs closely resemble those of APs found in other plant species [10,24], suggesting that the functions of these PeAPs have been evolutionarily preserved.
2.2. Phylogenetic Analysis of the PeAP Proteins
To investigate the phylogenetic connections among plant PeAPs, we created a dataset comprising 51 A. thaliana, 30 V. vinifera, and 55 P. euphratica AP amino acid sequences (Table S2), which were utilized to build a neighbor-joining tree. No assumptions were made regarding ancestral representation, relying solely on the relationships observed among the leaf nodes in unrooted trees. Consequently, the APs were categorized into three distinct groups (A, B, and C), as anticipated in Figure 2. The PeAP proteins demonstrated significant similarity to APs found in module plants. These genes were organized into clades alongside AtAPs and VvAPs, which exhibited superior bootstrap values. In the A clade, there were 5 AtAPs, 5 VvAPs, and 6 PeAPs, while the B group contained 4, 3, and 5 APs from each respective species. Notably, the C group included 44 PeAPs. This information suggests that the AP within the P. euphratica genome has undergone a distinct biological evolution when compared to those of the module plants. Across all species, the C group was the most populous, comprising 42 AtAPs, 22 VvAPs, and 44 PeAPs. In summary, our findings indicate that the APs present in the P. euphratica genome are highly conserved evolutionarily and contain numerous highly variable regions, making them particularly suitable for medical plant identification and systematic research.
2.3. Predicted Structure and Conserved Motifs Analysis of PeAP Proteins
To visualize the various protein structures of aspartic proteases, we selected representative proteins with similar amino acid lengths and compared their structures from each group of three plant species for analysis (Figure S1). Groups C proteins have similar structures among diverse plant species. What’s more, these proteins from groups A and B share significant overall structural similarity, certain regions exhibit distinct differences. Furthermore, the Group C protein sequences were highly conserved, although there were minor variations in the folding regions. Considering that the structure and characteristics of proteins influence the traits and roles of organisms, this information implies that the APs from these three plants could share homologous biological functions. The conserved roles of PeAPs indicate that they appear to be linked to particular functional guidelines.
To explore the evolutionary trajectory of the gene family, a comparison analysis of the gene structures of the PeAPs was conducted, as illustrated in Figure 3. Our examination of the genomic DNA sequences showed a variation in the number of introns, ranging from 0 to 13. The PeAPs predominantly exhibited highly comparable structures, which were categorized into Groups A and B branches within the NJ tree. All PeAPs belonging to Groups A and B displayed similar counts of introns and exons, except for Group C, which included 18 PeAPs that had lost introns. Additionally, to uncover potential motifs within the AP family of P. euphratica, we utilized the MEME website to predict the protein sequences of all complete PeAPs. As a result, we identified 10 distinct motifs from these proteins (Table S3). Apart from the identified motifs, amino acid sequence alignment of part of the sequences of abovementioned PeAPs (Figure S2). The group exhibited similar motifs, suggesting that these proteins may share certain common functions. In summary, this analysis reinforced the idea that motifs frequently found in aspartic proteases within the P. euphratica genome could be linked to the conserved functions of PeAPs, while the PeAPs appear to be connected to specific functional roles.
2.4. Evolutionary Relationships of AP Genes Between P. euphratica and Arabidopsis
In order to delve deeper into the origins and evolutionary development of P. euphratica AP genes, we examined the comparative synteny map that exists between the genomes of P. euphratica and Arabidopsis. As one of the key model plant species, Arabidopsis plays a significant role, especially concerning AP genes, as the functions of several of these genes have been thoroughly studied. Thus, through comparative genomics, we can determine the origin and diversification of P. euphratica APs based on their Arabidopsis homologs.
Large-scale syntenies containing 14 AP genes in populus and 20 in Arabidopsis were identified (Figure 4). In addition, 15 genes within the Arabidopsis genome, which were not classified as AP genes, were discovered to exhibit synteny with genes from grape P. euphratica (Figure 4 and Table S1). Concerning the individual correspondences between Populus and Arabidopsis AP genes, the syntenic relationships were clear-cut and comprised the following orthologous pairs: PeAP1-At3g54400, PeAP10-At3g61820, PeAP7-At1g77480, PeAP7-At1g44130, and PeAP10-At1g01300. This suggests that these genes likely existed in the genome of the most recent common ancestor shared by grape and Arabidopsis (Figure 4). The interpretation of synteny became more complex in instances where segmental duplications in populus aligned with just one Arabidopsis gene, or conversely, when a single gene from populus was associated with several Arabidopsis genes (Figure 4). The first situation was not detected, but the second included PeAP7-AT1G44130/AT1G77480, PeAP10-AT1G01300/AT3G61820, PeAP17-AT1G62300/AT4G04450, PeAP29-AT2G23950/AT4G30520, PeAP45-AT5G45130/AT4G19640, PeAP49-AT1G02500/AT4G01850 (Figure 4). Within this group, two orthologs of PeAP17 found in Arabidopsis (At1g62300, At4g04450) were not classified as AP genes. Nevertheless, both possess a WRKY domain, unlike PeAP17, which lacks the ASP domain. It is possible that PeAP17 has experienced several notable chromosomal rearrangements and fusions.
2.5. Subcellular Localization Prediction of PeAPs
Since details regarding subcellular locations can offer insights into predicting protein functions, our analysis suggests that all 22, 1, 2, 3, and 10 PeAPs are anticipated to be found in the chloroplast, extracellular space, nucleus, plasma membrane, and vacuole, respectively, with a high reliability index (RI > 6). However, exceptions were noted for PeAP2, PeAP7, PeAP11, PeAP14, PeAP18, PeAP25, PeAP29, PeAP31, PeAP32, PeAP35, PeAP44, PeAP47, PeAP53, PeAP54, PeAP55, PeAP57, and PeAP61, which may be situated in the nucleus, plasma membrane, and extracellular regions (Table S4). Notably, PeAP1 was exclusively found in the chloroplast (RI = 14), while the remaining PeAPs were predicted to be present in at least two different subcellular organelles (Table S4). The identified PeAP proteins in the P. euphratica genome exhibited various subcellular distributions, which showed functional diversity and may be involved in woody plant developmental growth.
2.6. Spatial and Temporal Expression of PeAPs in Various Developmental Tissues of P. euphratica Under Salt Treatment
To assess the transcriptional levels of PeAP genes across various organs, a heatmap utilizing RNA-seq data (Table S5), which were downloaded from the NCBI database (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA390611 (accessed on 1 March 2025)), was created. Our analysis revealed intricate, specific, and overlapping expressions of PeAPs in different tissues subjected to salt treatment. The organs were categorized into four distinct types: leaf, phloem, xylem, and root. The findings indicated that 55 PeAP genes exhibited unique expression patterns under varying salt stress conditions in these tissues. The transcriptional levels of these genes fluctuated among the different organs. For instance, 8 PeAPs (PeAP11, PeAP13, PeAP30, PeAP33, PeAP50, PeAP52, PeAP53, PeAP55) were found to be minimally detectable in whole plants; conversely, the transcriptional signals of 4 PeAP genes (PeAP9, PeAP10, PeAP22, PeAP49) were significantly prominent in the overall plantlet. Furthermore, transcript levels showed variability even within the same organs. In the four tissues of P. euphratica, the expression levels of the PeAP gene family members were generally low (Figure 5). Certain genes were exclusively expressed in specific tissues or organs; for example, PeAP48 was only detectable in the root. Overall, the PeAPs transcriptional levels differed among tissues and played a role in P. euphratica development.
2.7. Prediction of Cis-Acting Elements in the Promoter Regions of the PeAP Genes
To explore the possible functions of PeAPs, a database of plant promoters was utilized to locate the area situated 1.5 kb upstream from the transcription initiation site of PeAPs. In total, we discovered 14,488 transcription factor binding sites (TFBS) that fell into 93 distinct categories within the promoter regions of all PeAPs. The identified elements encompassed those related to stress, hormone responsiveness, light response, development, as well as promoter and enhancer functions, site-binding characteristics, and various other elements (Figure 6a). In comparison to other elements, there was a notable increase in the prevalence of hormone-responsive and stress-responsive elements (Figure 6a).
Populus euphratica exhibits remarkable stress tolerance in natural environments. TFBS analysis further revealed that the distribution abundance of stress-responsive TFBSs is significantly higher than other types of TFBSs (Figure 6a). Therefore, we initially focused on both biotic stress-related TFBSs and abiotic stress-related TFBSs in subsequent analyses. Within the various elements associated with biotic stress, the W-box, which is specifically identified by WRKY DNA binding proteins activated by salicylic acid (SA), exhibited a notable enrichment in the PeAPs promoter, surpassing other transcription factor binding sites related to biotic stress by over eight times. WB-box cis-element, which shows high similarity to W-box, also occurred in almost all PeAP promoters except for PeAP35. LS7, which acts as the positive salicylic acid-inducible element, is nearly ubiquitous across all PeAPs, with the exception of PeAP12 and AP31 (Figure 6b). SURECORE, as a sulfur starvation-responsive cis-element, exhibits an exceptionally high distribution density in the PeAP gene promoter region, significantly surpassing that of other cis-elements. PHR-1 binding site (P1BS), which accounts for phosphate starvation responses, occurred across approximately all promoter sequences of PeAPs except for PeAP55. MYB1, which is associated with dehydration-responsive genes, was distributed across all PeAPs and occurred more than 7 times in PeAP7, PeAP43, and PeAP49 (Figure 6c). Alongside the cis-elements mentioned earlier, ABRE elements categorized as hormone-related cis-elements were also widely identified within the promoter regions of PeAPs genes. These findings indicate that PeAPs could influence stress adaptability, responsiveness to phytohormones, and developmental growth.
2.8. Regulatory Network Mediated by P. euphratica AP Genes
Studying gene function is crucial for building networks that illustrate interactions among gene families. In this study, we developed a regulatory network governed by the aspartic protease genes of P. euphratica using STRING. Out of the 55 PeAPs analyzed, 19 APs were found to have significant interactions with other proteins, achieving a high confidence level (combined score > 0.5) (Figure S3). The other 9 interacted items correspond to 9 P. euphratica proteins listed in Table S6. Eight genes were detected as orthologues in Arabidopsis, except for PeuTF10G02367, without any orthologue in Arabidopsis in BLASTP results under a coverage > 60% threshold. Phylogenetic analysis supported that all conserved homologous genes in this cluster encode enzymes, with PeuTF05G00504 and PeuTF07G01029 specifically annotated as peroxidases (Figure S3). Their functional dominance suggests a potential role in mediating drought and osmotic stress tolerance in P. euphratica. All interacting proteins are enzymes, suggesting that aspartic protease may functionally cooperate with other enzymes.
3. Discussion
The gene family of aspartic proteases is crucial for how plants respond to stress and regulate their development [7]. In our research, we performed an extensive analysis across the genome of the AP gene family in Populus euphratica, a salt-tolerant woody species known for thriving in extreme saline conditions [25,26]. We identified 55 PeAP genes and classified them into three distinct groups based on conserved domain architecture. Our findings suggest that PeAPs have undergone significant evolutionary conservation while retaining functional plasticity, which may contribute to their roles in stress adaptation, particularly under saline conditions.
Phylogenetic analysis revealed that PeAPs cluster closely with their homologs in Arabidopsis thaliana and Vitis vinifera, indicating a conserved evolutionary trajectory among dicots [27]. However, PeAPs also exhibit lineage-specific expansions, particularly in clades associated with stress-responsive genes. This observation aligns with previous reports showing that gene family expansion in extremophytes like P. euphratica often correlates with enhanced abiotic stress tolerance [28,29,30]. Given that aspartic proteases are critical for stress memory in plants, the presence of PeAPs may imply a role in long-term salt adaptation.
Promoter analysis of PeAPs revealed an abundance of stress- and hormone-responsive cis-elements, including ABRE (abscisic acid-responsive), MYB (drought-inducible), and G-box (light-responsive) motifs. This finding suggests that PeAPs are tightly regulated by multiple signaling pathways, consistent with their putative roles in stress adaptation [18,26]. Notably, many PeAPs contain jasmonic acid (JA)- and salicylic acid (SA)-responsive elements, reinforcing the hypothesis that APs participate in defense responses [31]. Given that JA and SA pathways are crucial for salinity tolerance [32], the hormonal regulation of PeAPs may be a key factor in P. euphratica’s exceptional salt resilience.
Our expression profiling demonstrated that several PeAPs are significantly upregulated under salt stress, particularly AP12 and AP23. These genes share homology with Arabidopsis APs known to mediate osmotic stress responses [33]. Intriguingly, PeAP11 and PeAP49 exhibit structural similarity to CDR1 (Constitutive Disease Resistance 1), an AP involved in pathogen defense [34], suggesting a dual role in biotic and abiotic stress responses.
APs are known to degrade pathogenesis-related proteins, which can modulate stress-responsive pathways [35]. In P. euphratica, certain PeAPs may process pro-proteins involved in ion sequestration or reactive oxygen species (ROS) scavenging. For instance, PeAP27 contains a conserved domain similar to AtASP38 (AtPCS1) in A. thaliana, which regulates programmed cell death under oxidative stress [14]. Given that salinity induces ROS accumulation [36], PeAP-mediated protein processing may enhance antioxidant capacity, thereby improving salt tolerance.
Additionally, some PeAPs may influence cell wall remodeling, a critical process in salt adaptation. APs have been implicated in the deposition of secondary cell wall [37]. Since P. euphratica exhibits unique cell wall modifications under salt stress [38], PeAPs could contribute to maintaining structural integrity under osmotic stress. Unlike herbaceous models, woody plants possess a more complex AP gene family, likely due to their long-life cycles and perennial growth habits [37,38]. Our comparative analysis with A. thaliana APs revealed that PeAPs have undergone fewer tandem duplications but more transposon-mediated expansions, possibly reflecting adaptive evolution in extreme environments. This divergence suggests that P. euphratica has evolved specialized AP isoforms optimized for saline habitats. The identification of salt-responsive PeAPs provides promising candidates for improving salinity tolerance in sensitive crops. Overexpression of AP12 or AP23 in salt-sensitive poplar varieties could validate their functional roles. Given that APs are involved in multiple stress pathways [39], manipulating their expression may offer a multifaceted approach to enhancing abiotic stress resilience.
This research offers the initial comprehensive examination of the AP gene family within P. euphratica, emphasizing its possible function in adapting to saline environments. The conservation of stress-responsive cis-elements, coupled with the induction of specific PeAPs under salinity, underscores their importance in extremophyte biology. Future research should focus on functional characterization using CRISPR/Cas9-mediated knockout or overexpression studies to elucidate the precise mechanisms by which PeAPs confer salt tolerance.
4. Materials and Methods
4.1. Identification of AP Genes in Populus euphratica
A comprehensive analysis was conducted involving the retrieval of 51 Arabidopsis AP protein sequences. These sequences served as queries in a targeted search for potential AP genes within Populus euphratica reference genome [40]. This genome encompasses the chromosomal scale components associated with both male and female Populus euphratica, providing critical insights into the molecular mechanisms underlying gender determination and the phenomenon of sexual duality. The identification process was facilitated by the use of BLASTP (v2.15.0) software, which enabled the researchers to effectively pinpoint and analyze the relevant genetic sequences. Then, HMMER (v3.3.1) program and PFAM (PF00026) were employed to identify candidate AP protein in P. euphratica. The intersection of genes from both methods was treated as a highly confidential candidate P. euphratica AP genes. Protein sequences of all candidate AP genes were sent to NCBI-CDD to confirm that they have the complete Asp domain [41]. The molecular weight, hydrophobic characteristics, and isoelectric point were determined using an R package Peptides (v2.4.6).
4.2. Multiple Sequence Alignment and Phylogenetic Analysis
A total of six Populus euphratica candidate aspartic protease (AP) proteins lacking complete ASP domains were excluded from phylogenetic analysis and subsequent investigations. The remaining 55 PeAPs, along with 30 Vitis vinifera APs (VvAPs), 51 Arabidopsis thaliana APs (AtAPs; pepsin-like type), barley nucellins (GenBank accession no. AAB96882.1), tobacco CND41 (BAA22813.1), cardoon cardosin A (CAB40134), and porcine pepsin A (NP_999038.2), were subjected to multiple sequence alignment using MAFFT. Conserved sites were subsequently extracted from the alignment using Gblocks (v0.91b) [42] to remove divergent and ambiguously aligned regions. Phylogenetic reconstruction was performed with IQ-TREE (v2.2.0) employing the Neighbor-Joining (NJ) method under default parameters. The resulting phylogenetic trees were visualized and annotated using iTOL [43], with color-coding applied according to established aspartic protease classification groups.
4.3. Gene and Protein Structure Analysis
Genomic sequences along with their associated coding sequences (CDSs) for PeAPs were obtained from the genome of Populus euphratica utilizing SeqKit and TBtools. In order to discover conserved motifs within PeAP proteins, we utilized the Multiple Expectation Maximization for Motif Elucidation (MEME) suite (v5.2.0), applying these parameters: Optimal motif width ranging from 10 to 50 amino acids; Maximum number of motifs set to 10; All other configurations were kept at their default settings. For each PeAP protein, functional domains were predicted and annotated using NCBI’s Conserved Domain Database (CDD) and SMART (Simple Modular Architecture Research Tool). Finally, schematic representations of protein structures were generated using TBtools.
4.4. Cis-Elements Identification
The promoter sequences of PeAP genes, measuring 1.5 kb and located upstream of the TSS site, were extracted by bedtools from the genome sequence according to the position record in genome annotation file. Promoter sequences were submitted to PlantPAN 4.0 web tool for TFBS identification. All identified TFBS were further classified into 6 groups as stress-related, hormone-responsive related, light-responsive, development-related, promoter/enhancer elements, and others according to TFBS functional annotations. All stress-related cis-elements were further validated by manual inspection. The cis-elements distribution visualized by TBtools.
4.5. Analysis of Transcriptional Profiles
The sequencing data for the transcriptome of Populus euphratica were sourced from the NCBI Sequence Read Archive (SRA) with the accession number SRP116293. The complementary DNA (cDNA) libraries derived from both control and salt-stressed samples of leaf, phloem, xylem, and root tissues have the following accession numbers: SRX3139499, SRX3139976, SRX3139977, and SRX3140050, respectively. Initially, the raw sequencing reads were evaluated for quality using FastQC (v0.11.9) and subsequently processed through fastp (v0.23.2) utilizing default settings for adapter trimming and quality filtering. The STAR aligner (v2.7.10b) was utilized with default parameters to align high-quality reads to the reference genome of Populus euphratica (female). Gene expression quantification was performed using feature Counts (v2.0.3) to generate read counts for all annotated genes across each sample.
4.6. Analyses of Synteny in PeAPs
Information regarding the physical locations of the PeAP genes was obtained from the genome database of P. euphratica. To examine the homology of AP genes between P. euphratica and A. thaliana, the MCScanX program was utilized with its default settings. All synteny information about P. euphratica and A. thaliana was visualized by TBtools.
4.7. Prediction of 3-Dimensional Structures and Interaction Network of PeAP Proteins
The three-dimensional (3D) configurations of PeAP proteins were visualized using the SWISS-MODEL database, from which 3D diagrams were obtained. A 3D protein model was constructed with complete confidence for all genes that were positively predicted, while the coverage of residues ranged between 78% and 98%. The 3D structures from each group were randomly selected to make a comparison among A. thaliana, V. vinifera, and P. euphratica. Moreover, we created a protein–protein interaction network of 55 PeAP proteins based on their homologs in Populus trichocarpa versus A. thaliana using the STRING online database (https://string-db.org, accessed on 20 April 2025).
5. Conclusions
This research presents the initial comprehensive examination of the Aspartic protease (AP) gene family within Populus euphratica, revealing 45 PeAP genes that may contribute to salt tolerance. Phylogenetic and expression analyses revealed that several PeAPs (e.g., PeAP22 and PeAP51) are strongly induced by salt stress and may function in stress adaptation through protein processing, ROS scavenging, or cell wall remodeling. The findings offer valuable genetic resources for improving salt tolerance in woody crops through molecular breeding. Future work may involve CRISPR-Cas9 knockout validation of PeAP22 and PeAP51 to dissect their functional roles in ionic homeostasis or ROS detoxification.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Shrivastava P. Kumar R. Soil salinity: A serious environmental issue and plant growth promoting bacteria as one of the tools for its alleviation Saudi J. Biol. Sci.20152212313110.1016/j.sjbs.2014.12.00125737642 PMC 4336437 · doi ↗ · pubmed ↗
- 2Arif Y. Singh P. Siddiqui H. Bajguz A. Hayat S. Salinity induced physiological and biochemical changes in plants: An omic approach towards salt stress tolerance Plant Physiol. Biochem.2020156647710.1016/j.plaphy.2020.08.04232906023 · doi ↗ · pubmed ↗
- 3Balasubramaniam T. Shen G. Esmaeili N. Zhang H. Plants’ Response Mechanisms to Salinity Stress Plants 202312225310.3390/plants 1212225337375879 PMC 10300796 · doi ↗ · pubmed ↗
- 4Chen S. Hawighorst P. Sun J. Polle A. Salt tolerance in Populus: Significance of stress signaling networks, mycorrhization, and soil amendments for cellular and whole-plant nutrition Environ. Exp. Bot.201410711312410.1016/j.envexpbot.2014.06.001 · doi ↗
- 5Watanabe S. Kojima K. Ide Y. Sasaki S. Effects of saline and osmotic stress on proline and sugar accumulation in Populus euphratica in vitro Plant Cell Tissue Organ Cult. (PCTOC)20006319920610.1023/A:1010619503680 · doi ↗
- 6Soares A. Ribeiro Carlton S.M. Simões I. Atypical and nucellin-like aspartic proteases: Emerging players in plant developmental processes and stress responses J. Exp. Bot.2019702059207610.1093/jxb/erz 03430715463 · doi ↗ · pubmed ↗
- 7Cao S. Guo M. Wang C. Xu W. Shi T. Tong G. Zhen C. Cheng H. Yang C. Elsheery N.I. Genome-wide characterization of aspartic protease (AP) gene family in Populus trichocarpa and identification of the potential Pt A Ps involved in wood formation BMC Plant Biol.20191927610.1186/s 12870-019-1865-031234799 PMC 6591973 · doi ↗ · pubmed ↗
- 8Simoes I. Faro C. Structure and function of plant aspartic proteinases Eur. J. Biochem.20042712067207510.1111/j.1432-1033.2004.04136.x 15153096 · doi ↗ · pubmed ↗