Comparative Analysis of Chloroplast Genomes of 19 Saxifraga Species, Mostly from the European Alps
Zhenning Leng, Zhe Pang, Zaijun He, Qingbo Gao

TL;DR
This study analyzes chloroplast genomes of 19 Saxifraga species, mostly from the European Alps, to better understand their genetic diversity and evolutionary relationships.
Contribution
The study provides the first comprehensive chloroplast genome data for 15 Saxifraga species from the European Alps, enabling broader comparative and evolutionary analyses.
Findings
Chloroplast genomes of 19 Saxifraga species were sequenced, revealing a typical quadripartite structure and 113 unique genes per genome.
Six intergenic regions were identified as potential DNA barcodes for molecular marker studies.
Phylogenetic analysis using 75 protein-coding genes showed high bootstrap support and a topology consistent with prior research.
Abstract
Complete chloroplast genome sequences are widely used in the analyses of phylogenetic relationships among angiosperms. As a species-rich genus, species diversity centers of Saxifraga L. include mountainous regions of Eurasia, such as the Alps and the Qinghai–Tibetan Plateau (QTP) sensu lato. However, to date, datasets of chloroplast genomes of Saxifraga have been concentrated on the QTP species; those from European Alps are largely unavailable, which hinders comprehensively comparative and evolutionary analyses of chloroplast genomes in this genus. Here, complete chloroplast genomes of 19 Saxifraga species were de novo sequenced, assembled and annotated, and of these 15 species from Alps were reported for the first time. Subsequent comparative analysis and phylogenetic reconstruction were also conducted. Chloroplast genome length of the 19 Saxifraga species range from 149,217 bp to…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
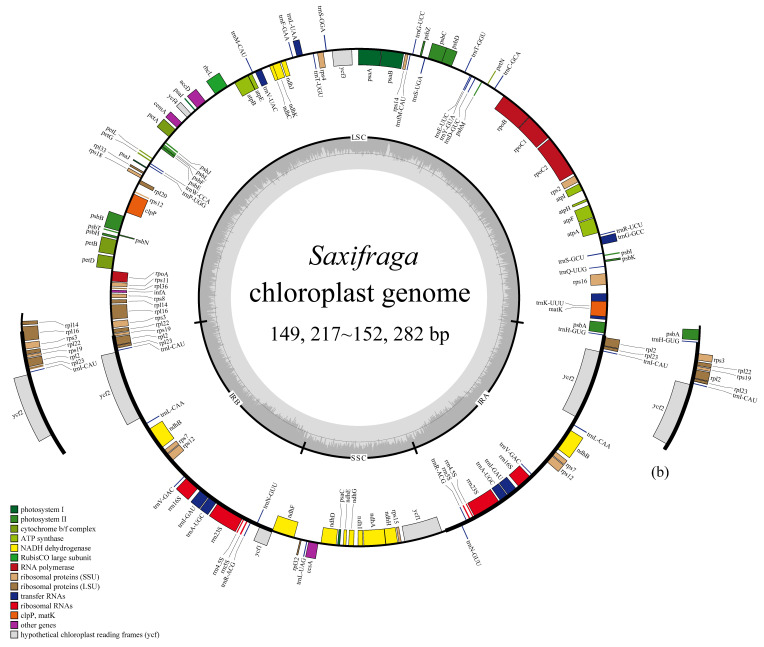 Figure 1
Figure 1 Figure 2
Figure 2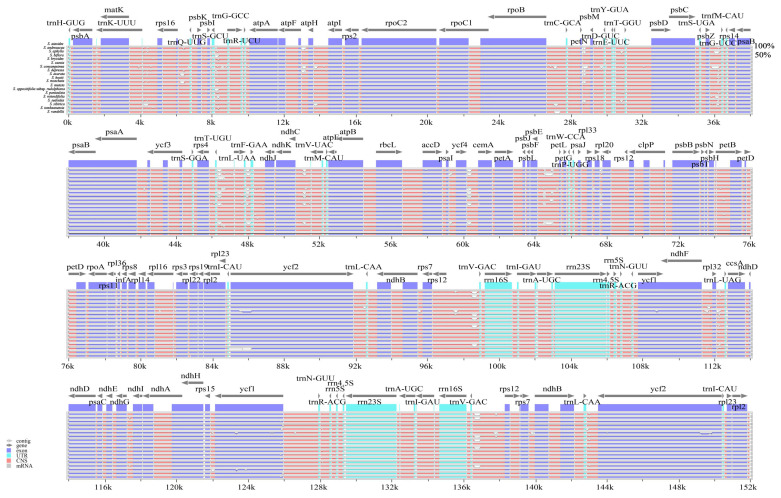 Figure 3
Figure 3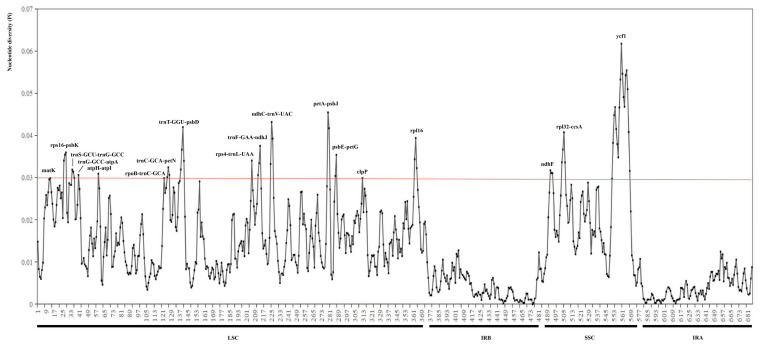 Figure 4
Figure 4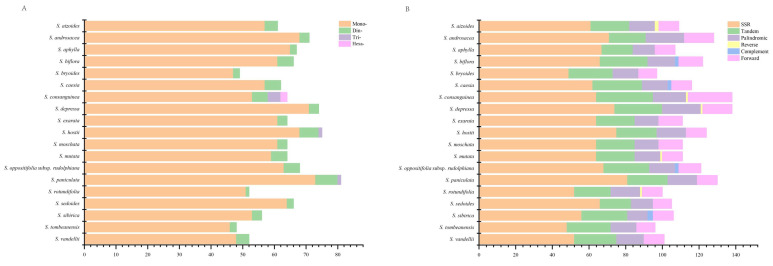 Figure 5
Figure 5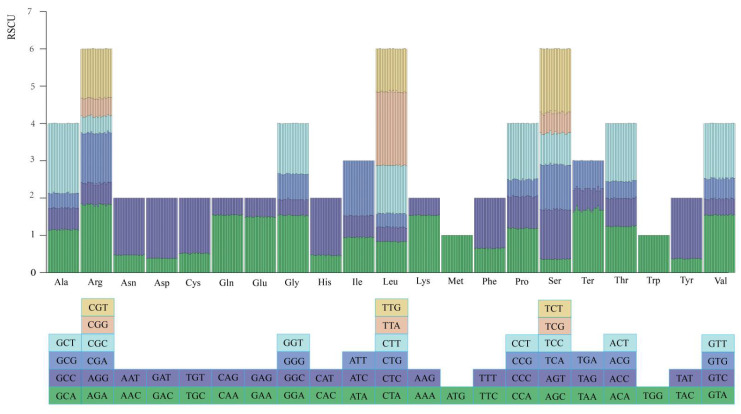 Figure 6
Figure 6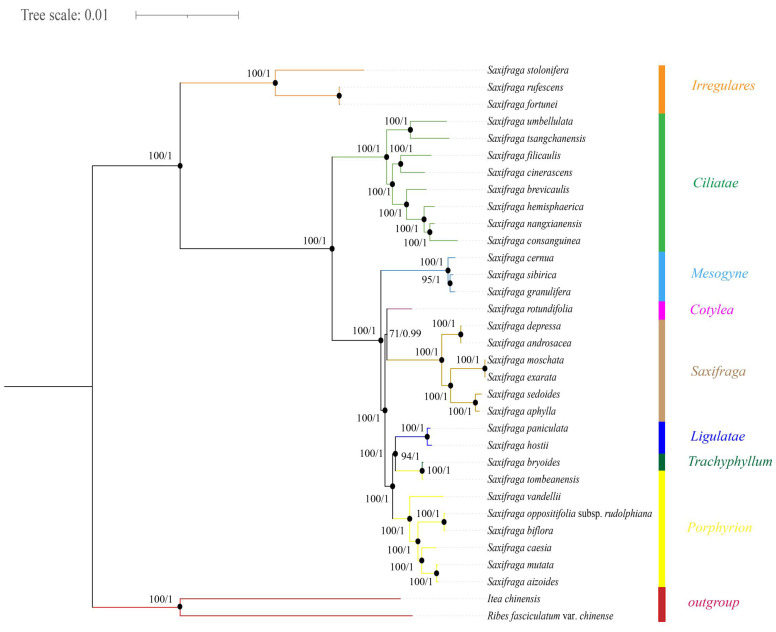 Figure 7
Figure 7- —Qinghai Provincial Science and Technology Major Project
- —Chinese Academy of Sciences (CAS) International Innovation Team
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Protist diversity and phylogeny · Plant Pathogens and Fungal Diseases
1. Introduction
The large arctic–alpine genus Saxifraga L. (Saxifragaceae), which comprises 450–500 species, is widely distributed across the Northern Hemisphere [1]. At least two mountainous regions can be recognized as species diversity centers of Saxifraga: the Alps in Europe [2] and the Qinghai–Tibetan Plateau sensu lato (including Himalayas, Henduan Mts., and plateau platform) in Asia [3]. Considering considerable species richness and habitat diversity, as well as a corresponding high level of morphological, physiological and life cycle diversity [4], Saxifraga offers an ideal system to reveal potential driving factors for the formation of high levels of biodiversity in association with mountains, as in [1] or [5,6,7].
A finely resolved and well supported phylogenetic topology of a given taxonomic group is essential to conduct further evolutionary studies, such as biogeographic analysis, trait reconstruction and ploidy evolution [8]. However, phylogenetic relationships of lineages which have experienced recent rapid radiation could not be well resolved by traditionally universal DNA markers [9]. As for the phylogeny of Saxifraga, although the infrageneric skeleton frame at sectional level has been revealed to be based on two universal markers [10], phylogenetic relationships within sections were not well resolved [4,10], partly due to rapid radiation [1]. Because of the small size, relatively conservative structure and maternal inheritance, chloroplast genomes have been widely applied to plant phylogeny and evolution, e.g., in [11,12,13]. To date, the large chloroplast genome dataset of Saxifraga has been predominantly focused on S. sect. Ciliatae Haw. [14], the most species-rich section whose center of diversity is the QTP sensu lato. The phylogenetic relationship was well resolved within this recent-radiation section, indicating a potentially good performance of chloroplast genome data on the reconstruction of phylogenetic relationships at an infra-sectional level of Saxifraga. However, chloroplast genomes from the remaining sections are limited, even absent, especially those from the Alps, which hinders reliable phylogenetic reconstructions both at generic and infra-sectional levels. On the other hand, although the structure and gene content of chloroplast genomes are conserved in most angiosperms, extensive gene losses and large inversions have been detected in several lineages, such as Gentianaceae [15,16], Asteraceae [17,18] and Leguminosae [19,20]. Previous studies [9,14] have confirmed high conservation in structural organization, gene arrangement and gene content in chloroplast genomes of S. sect. Ciliatae. However, whether this is the case at the generic level of Saxifraga is still unclear due to the asymmetry in the available data of chloroplast genomes between the QTP and Alps.
In this study, 19 chloroplast genomes in Saxifraga were de novo sequenced, assembled and annotated, representing 7 of the 13 sections [10]. Among the 19 species included in this study, 17 are from the European Alps and 15 of which are sequenced for the first time. Comparative analyses were conducted to reveal the molecular evolution of chloroplast genomes in Saxifraga. Meanwhile, the performance of phylogenetic reconstruction was tested based on the 19 newly generated sequences combined with additional 12 Saxifraga species downloaded from NCBI. This study will enlarge the dataset of Saxifraga chloroplast genomes, improve the recognition of chloroplast genome structure and provide a general phylogeny outline of the whole genus.
2. Results
2.1. Comparative Chloroplast Genomes of Saxifraga Species
2.1.1. General Feature Comparison
The chloroplast genomes of the 19 Saxifraga species have a typical quadripartite structure and contain a large single copy (LSC), a small single copy (SSC), and two copies of inverted repeat (IR) regions (Figure 1). The size of the 19 chloroplast genomes ranges from 149,217 bp (S. aphylla Sternb.) to 152,282 bp (S. rotundifolia L.) (Table 1). Saxifraga consanguinea W. W. Sm. has the largest IR region (27,220 bp), but the smallest LSC (78,948) and SSC (16,659) among the 19 chloroplast genomes. The largest LSC (83,618) and SSC (17,352) occur in S. rotundifolia, while the smallest IR (25,378) occurs in S. androsacea L. (Table 1). The total GC content varies from 37.66% to 37.84%, and GC contents of LSC, SSC and IR range from 35.67% to 36.16%, 31.70% to 32.10% and 42.04% to 42.92%, respectively (Table 1). Despite size variations, the GC-contents are similar among the 19 Saxifraga species in the whole genome, LSC, SSC and IR regions.
Each of the 19 newly generated Saxifraga chloroplast genomes contains 113 unique genes, among which 79 genes are PCGs, 4 are rRNA genes and 30 are tRNA genes (Table 2). Among the 113 unique genes, 7 PCGs (ndhB, rpl2, rpl23, rps12, rps7, ycf2, ycf1), all the 4 rRNA genes and 7 tRNA genes (trnA-UGC, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG) are completely or partially duplicated in the IR region, which results in a total number of 131 genes in all of the remaining 18 genomes except S. consanguinea (Table 2). As for the chloroplast genome of S. consanguinea, 3 additional PCGs (rps19, rpl22, rps3) are also duplicated in the IR region, leading to a total number of 134 genes in this species. By counting the introns in genes, 2 genes have two introns and 15 genes have one intron (Table 2). The rps12 gene is trans-spliced, with its 5′-end exon located in the LSC region and 3′-end exon duplicated in the IRs. Regarding the function of genes, 45 of them take part in photosynthesis, 59 in self-replication and 9 in other functions (Table 2).
2.1.2. IR Boundary Variation Analysis
The boundaries between LSC, IR and SSC, as well as the adjacent genes, were compared across the 19 Saxifraga chloroplast genomes (Figure 2). The most massive expansion of IR boundaries was detected between LSC and IRB regions in S. consanguinea, which expended to the rpl16 gene in LSC. This resulted in a duplication of three additional PCGs (rps19, rpl22, rps3) compared to the remaining chloroplast genomes, leading to the largest IR region (27,220 bp), but the smallest LSC (78,948) and SSC (16,659), in S. consanguinea (Table 1, Figure 2). However, for the remaining 18 chloroplast genomes, the LSC/IRB junctions were all located within the coding region of rps19. The expansion/contraction of IRs were also detected in junctions of IRB/SSC which fell into the ycf1 pseudogene and/or ndhF gene, as well as SSC/IRA which was located within the ycf1 gene but with different extensions (Figure 2). The IRA/LSC border of the 19 chloroplast genomes was mainly conserved, exactly matching or being a few basepairs ahead of the start codon of trnH (Figure 2).
2.1.3. Polymorphic Analysis Among Chloroplast Genome Sequences
The overall sequence identity of the 19 chloroplast genomes of Saxifraga was plotted using mVISTA with the annotation of the S. aizoides L. chloroplast genome as the reference (Figure 3). The results showed that the Saxifraga chloroplast genomes exhibited a high level of sequence synteny, suggesting a conserved evolutionary pattern. Nucleotide variability values were calculated using the window sliding analysis, as implemented in DnaSP (Figure 4). A total of 685 highly variable regions were identified among the 19 chloroplast genomes, of which 18 regions exhibited a nucleotide diversity value (π) higher than 0.03. As a whole, the IR regions are less divergent compared to the LSC and SSC region, and the coding regions are more conserved than the intergenic spacers and introns. Among the 18 most variable regions, 13 locations (rps16-psbK, trnS-GCU-trnG-GCC, trnG-GCC-atpA, atpH-atpI, rpoB-trnC-GCA, trnC-GCA-petN, trnT-GGU-psbD, rps4-trnL-UAA, trnF-GAA-ndhJ, ndhC-trnV-UAC, petA-psbJ, psbE-petG, rpl32-ccsA) are intergenic regions, and 5 locations (matK, clpP, rpl16, ndhF, ycf1) are protein coding regions. The ycf1 pseudogene, which is located at the boundary of SSC/IRA, had the highest nucleotide variation (π = 0.06174). Its high polymorphism may relate to the expansion/contraction of IR regions. These highly variable regions can be used as candidates of the DNA barcodes for Saxifraga.
2.1.4. Repeat Sequences Analysis
A total of 1204 SSRs were identified among the 19 Saxifraga chloroplast genomes. The mono-, di-, tri- and hexa-nucleotide repeats accounted for 93.11%, 5.82%, 0.77% and 0.31%, respectively. No tetra- or penta-nucleotide repeats were detected in the 19 Saxifraga chloroplast genomes included in this study (Figure 5). The two dominant SSRs motif types were A/T and AT/TA. S. paniculata Mill. possessed the highest number of SSRs, while S. consanguinea had the highest abundance of SSRs types (Figure 5). A total of 433 Tandem repeats and 534 large sequence repeats (LSRs; ≥30 bp and Hamming distance = 3) were identified among the 19 Saxifraga chloroplast genomes. Both of these were most abundant in the S. consanguinea chloroplast genome compared to the remaining genomes (Figure 5).
2.1.5. Codon Usage Analysis
Condon usage analysis revealed that the 19 Saxifraga species contained 61 codons encoding 20 amino acids, as well as three stop codons (Figure 6). The total number of codons of the 19 species ranged from 19,897 in S. sedoides L. to 20,709 in S. rotundifolia L. Leucine is the most frequently found amino acid (40,377), and cysteine is the rarest (4382). The RSCU value calculation result showed 30 codons with an RSCU value greater than 1, suggesting a higher codon usage frequency than expected, while there were 32 codons with an RSCU value less than 1, indicating a lower codon usage frequency. Among the codons with RSCU value > 1, 12 codons ended with A, 16 codons ended with T, and only 1 codon ended with G, indicating an A/T ending preference of Saxifraga chloroplast genome codons.
2.2. Phylogenetic Analysis
In total, 75 common genes (accD, atpA, atpB, atpE, atpF, atpH, atpI, ccsA, cemA, infA, matK, ndhA, ndhB, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK, petA, petB, petD, petG, petL, petN, psaA, psaB, psaC, psaI, psaJ, psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbT, psbZ, rbcL, rpl2, rpl14, rpl16, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36, rpoA, rpoB, rpoC1, rpoC2, rps2, rps3, rps4, rps7, rps8, rps11, rps12, rps14, rps15, rps16, rps18, rps19, ycf1, ycf2) were extracted, and phylogenetic relationships were reconstructed using R. fasciculatum var. chinense and I. chinensis as outgroups. Referring to the description of phylogenetic results by Tkach [10], firstly, three species—S. stolonifera, S. rufescens and S. fortunei—were clustered together, belonging to sect. Irregulares. Sect. Ciliatae consisted of eight species, including S. umbellulata, S. tsangchanensis, S. filicaulis, S. cinerascens, S. brevicaulis, S. hemisphaerica, S. nangxianensis and S. consanguinea. Sect. Mesogyne included S. cernua, S. sibirica and S. granulifera, while Sect. Cotylea was represented solely by S. rotundifolia. Six species, including S. depressa, S. androsacea, S. moschata, S. exarata, S. sedoides and S. aphylla, were assigned to sect. Saxifraga. Sect. Ligulatae contained S. paniculata and S. hostii. Sect. Trachyphyllum was represented by S. bryoides. Sect. Porphyrion encompassed seven species, including S. tombeanensis, S. vandellii, S. oppositifolia subsp. rudolphiana, S. biflora, S. caesia, S. mutata and S.aizoides. The ML and BI tree topologies were highly congruent and were consistent with previous studies [10]. The BS and PP values were fairly high, all but three nodes presented a BS value of 100% and all nodes of PP values reached 1, with the exception of one node (Figure 7).
3. Discussion
3.1. Chloroplast Genome Structure Variation Within the 19 Saxifraga Species
In general, the gene content and gene organization of angiosperm chloroplast genomes are highly conserved compared to nuclear and mitochondrial genomes [21]. Because of the high stability and conservatism, the whole chloroplast genome has been widely used in plant species identification, population genetics, genome evolution and phylogenetic studies [22]. However, the phenomena of gene rearrangement [17], large fragment loss [16] and even structural variations [23] still exist in some lineages. In this study, the complete chloroplast genomes of 19 Saxifraga species were de novo sequenced and analyzed. Among them, 15 species from the European Alps have been sequenced for the first time. The genome structure of the 19 Saxifraga species is consistent with those of most terrestrial plants, and the size (ranging from 149,217 bp to 152,282 bp) falls well into the range of 120–160 kb of angiosperm chloroplast genomes [24]. The GC contents of the 19 chloroplast genome sequences (37.66–37.84%) were similar to the average of sequenced land plants (37.6%) [25], and the IR regions contain the highest GC content, which is in line with most angiosperm plants. Structure analyses of these newly generated chloroplast genomes show high conservation in structural organization, gene arrangement and gene content. Large structural variation [23], large fragments inversion/loss [16,17] and gene rearrangement [26] are not detected in chloroplast genomes generated in this study, which is congruent with previous studies [9,14]. To date, the largest chloroplast genome dataset of Saxifraga was focused on sect. Ciliatae, whose diversity center is the QTP sensu lato [14]. Nearly one hundred chloroplast genomes are available for this largest section of Saxifraga, and genome structure has been confirmed to be rather conservative [14]. In this study, chloroplast genomes of 15 Saxifraga species from the Alps were generated for the first time, which can give us a relatively comprehensive scope of chloroplast genome evolution at the generic level of Saxifraga. Although chloroplast genome data of European species are still urgently needed, our results, combined with those published data, may suggest a relatively conservative evolutionary history of chloroplast genomes at the generic level of Saxifraga.
The expansion and contraction of the chloroplast genome is a common evolutionary phenomenon in plants [27]. In angiosperms, the expansion/contraction of the IR boundaries of chloroplast genomes often result in different levels of genome size variation, gene duplication or production of pseudogenes [28,29,30]. Because of the expansion of the IR boundary, S. consanguinea has the biggest length in IR regions among 19 Saxifraga species. The large IRs of the plastomes are hypothesized to contribute to plastome stabilization because their absence often coincides with severe changes in gene order [31]. However, the SSC and LSC regions are the smallest of S. consanguinea, and kept the total length relatively consistent. The balance between the SC regions and IR regions might be the reason for the stability in length. Although an extension of IR regions was detected in S. consanguinea, it seems that IR boundaries are much conservative among Saxifraga species: 18 out of the 19 chloroplast genomes share the same type of IR boundaries. This was also revealed by Yuan et al. [14], in which 88 of the 94 sect. Ciliatae chloroplast genomes shared the same type of IR boundaries, and only 6 species showed IR expansion/contraction. Pseudogenes play an essential role in gene expression regulation and genome evolution [32]. Two pseudogenes of rps19 and ycf1 were found in this study, coincident with the results revealed in Aconitum [33]. Meanwhile, pseudogene rps19 was found in 18 of the 19 Saxifraga species, while ycf1 was detected in 8 species, indicating a species specificity of pseudogenes among species. Meanwhile, ycf1 has been confirmed to be associated with high altitude [34], and so the duplication in IR regions might contribute to its adaptation.
Although chloroplast genomes in this study exhibit high conservation in genome structure and IR boundary, regions with high sequence polymorphisms (e.g., ndhC-trnV, psbE-petL, rpl32-trnL, rps16-trnQ, trnF-ndhJ, trnS-trnG, ycf1) are observed among the 19 chloroplast genomes. These highly divergent regions are also revealed at the section level of sect. Ciliatae [14], as well as at the family level of Saxifragaceae [9], and can be used as candidate barcoding regions for species identification, population genetics and phylogenetics of Saxifraga.
Due to high levels of variations, chloroplast SSRs (≥10 bp) play an important role in polymorphism investigations, population genetics and phylogenetic analyses [35,36,37,38,39]. In this study, the number of SSRs ranges from 48 to 81 among the 19 Saxifraga species, which shows a moderate level compared with other species of angiosperms [17,40,41]. Most of these SSRs are located in the LSC region, followed by the SSC and IR regions. The high level of A/T content and the predominance of mononucleotide repeats are the significant features in chloroplast SSRs of the 19 Saxifraga species, which may reflect a common phenomenon in angiosperms [15,42]. In addition, the high AT content of chloroplast SSRs may be caused by the main contribution of poly (A), poly (T) or poly (AT) repeats in the non-coding regions of the single-copy regions, especially in the LSC region [43]. Furthermore, SSRs detected in this study are mainly distributed in the non-coding region, including intergenetic regions and gene introns. In summary, the range of SSR numbers, frequency of different SSRs types and the distribution patterns of SSRs across the 19 chloroplast genomes are comparable with those in sect. Ciliatae [9,14]. Long repeats play an essential role in the whole-chloroplast genome variation, expansion and rearrangement [44]. We identified 17–31 tandem repeats, as well as 22–43 large sequence repeats, among the 19 chloroplast genomes, indicating a relative abundance of long repeats in Saxifraga chloroplast genomes. These SSRs and long repeats usually exhibit high levels of variation, and thus provide potential candidates for the development of molecular markers for future evolutionary and genetic diversity studies in Saxifraga.
Codon usage preference has been documented as one of the evolutionary features in many organisms [45]. Our results revealed that the number of codon types and the frequency of amino acids, as well as codon usage preference, are similar to those revealed in sect. Ciliatae [14].
Despite the limited number of species included in this study, it represents 7 of the 13 sections of Saxifraga [10]. According to our results in this study, together with previous studies [9,14,46], chloroplast genomes of Saxifraga may have experienced a conservative evolutionary history, as proven by (i) high conservation in structural organization, gene arrangement and gene content; (ii) many conservative IR boundaries; (iii) similarity in SSRs numbers, types frequencies and distribution patterns; and (iv) comparable codon types and codon usage preference. However, more chloroplast genomes of Saxifraga species, especially those from the European Alps, are needed to test a comprehensive chloroplast genome evolution in this genus.
3.2. Phylogeny of Saxifraga Species
To date, the most comprehensive investigation on the phylogenetic relationships at the generic level of Saxifraga was that conducted by Tkach et al. [10], employing a balanced sampling strategy of 254 Saxifraga species and two universal DNA markers (nrDNA ITS and plastid trnL-F). A backbone framework of at least 13 sections and 9 subsections was recognized within Saxifraga [10]. However, due to the recent rapid radiation of this species-rich genus [1], some difficulties may occur during phylogenetic reconstruction when using only a few universal DNA markers. On the one hand, rapid radiations are usually associated with low genetic differentiation between closely related clades or species, making phylogenetic reconstruction difficult. On the other hand, limited informative sites when using only a few DNA markers might not reveal a well-resolved phylogenetic relationship. This is the case in the phylogenetic study of Saxifraga at the moment. Firstly, some of the major clades within Saxifraga as recognized by Tkach et al. [10] are not well supported, leading to an unclear placement or relationship on the phylogenetic tree. Secondly, the phylogenetic relationships at the infra-section/subsection level are not well resolved, especially for those with high species richness, such as sect. Saxifraga and sect. Porphyrion Tausch. Thirdly, taxonomic positions of some Saxifraga species, such as S. odontophylla Wall. ex Sternb. and S. nana Engl., are still ambiguous. Complete chloroplast genome sequences seem to offer an opportunity to resolve problems in Saxifraga phylogenetic study, as mentioned above. Yuan et al. [14] employed ca. 100 chloroplast genomes to investigate the infra-section relationships of sect. Ciliatae, the most species-rich section which has experienced recent radiation. Phylogenetic relationships within sect. Ciliatae using complete chloroplast genome sequences are better resolved and have higher support values compared to that using few DNA markers [4]. In this study, 31 Saxifraga species from eight sections are employed to test the performance of the complete chloroplast genome on the resolution of phylogenetic relationships of Saxifraga. The resolution and support values as revealed by phylogenetic tree are extremely high even at the tip nodes, suggesting a good performance of complete chloroplast genome sequences on the resolution of phylogenetic relationships of Saxifraga at infra-section/subsection level.
In conclusion, our study adds new complete chloroplast genome sequences to the molecular dataset of Saxifraga. The evolutionary history of chloroplast genomes of Saxifraga may be conservative. Complete chloroplast genome sequences seem to offer an opportunity to resolve phylogenetic relationships at the infra-section/subsection level of Saxifraga, but more new sequences should be generated.
4. Materials and Methods
4.1. Sample Collection, DNA Extraction, and Sequencing
A total of 19 species were sampled, of which 17 were from the Alps and 2 from Sino-Himalaya (Table 3). The 19 species represent 7 of the 13 sections proposed by Gornall [47] and Tkach et al. [10]. Leaves were collected from a single individual, then dried in silica gel. The voucher specimen was deposited in the herbarium of the University of Leicester, Leicester, England, and the Northwest Institute of Plateau Biology (HNWP), Xining, Qinghai, China. Total genomic DNA was extracted from silica-dried leaves by the DNA quick extraction system (DP321) according to the manufacturer’s protocol (Tiangen Biochemical Technology Co., Ltd., Beijing, China). Total DNA was then randomly fragmented with the Covaris ultrasonic crusher. A series of steps was performed to complete library construction, such as end repair and phosphorylation, a-tail addition, sequencing connector addition, purification and PCR amplification. Finally, the qualified libraries were pooled into flowcells. Paired-end sequencing from both ends of 150 bp fragments was performed on the Illumina NovaSeq 6000 platform at the BENAGEN company in Wuhan, China, to generate ~5 Gb data for each individual. We used fastp v.0.23.1 [48] to filter raw sequence reads when (i) the N content in any read was more than 10% of the base; (ii) the number of low-quality (Q ≤ 5) bases in any read exceeded 50%; and (iii) any read contained the adapter content [49].
4.2. Chloroplast Genome Assembly and Annotation
High-quality clean reads were assembled using GetOrganelle v.1.7.5 [50] with the default parameters and S. sinomontana J-T. Pan & Gornall as a reference genome (GenBank accession no. MN104589) [9]. Circular genomes were annotated using CPGAVAS2 (http://47.96.249.172:16019/analyzer/home (accessed on 22 January 2025)) [51]. The preliminarily annotation results may have some problems, such as genes with nonstandard start or stop codons or genes with an internal stop codon. The correctness was checked using CPGView [52]. Finally, well-annotated sequences were then submitted to OGDRAW (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html (accessed on 20 December 2024)) for chloroplast genome visualization [53]. All the complete chloroplast genome sequences were deposited into GenBank, with the accession numbers PV423509 and PV426729-426746.
4.3. Comparative Analysis of Chloroplast Genomes
The length, GC content of the total sequence, LSC region, SSC region and IR regions, as well as numbers of protein-coding genes, tRNA genes and rRNA genes, were calculated by command lines using Qt Console v5.5.1. The gene types were counted by CPStools [54]. A comparison of junction sites of LSC, IR and SSC regions was implemented using the program IRscope (http://msgvd.genehub.com.cn/IRscope/ (accessed on 27 November 2024)) [55]. The percentage of sequence identity was analyzed and plotted using the program mVISTA (https://genome.lbl.gov/vista/mvista/submit.shtml (accessed on 18 December 2024)) [56], with an alignment algorithm of LAGAN. To identify high variable regions, the 19 chloroplast genomes were aligned using MAFFT v.7 [57] in PhyloSuite [58] with default parameters. The number of polymorphic sites and nucleotide variability (Pi) were evaluated using a sliding window with 200 bp step size and a 600 bp window length implemented in DnaSP v5.10.1 [59]. Simple sequence repeats (SSRs) were detected using MISA (https://webblast.ipk-gatersleben.de/misa/ (accessed on 8 January 2025)) [60]. Tandem repeats were identified with Tandem Repeats Finder v4.09 [61]. Large sequence repeats (LSRs) were identified using REPuter with hamming distance = 3, maximum computed repeats of 50 bp and minimum repeat size of 30 bp [62]. The codon usage and the relative synonymous codon usage (RSCU) value were estimated using CPStools [54]. To reduce sampling error, protein-coding genes (PCGs) shorter than 300 bp and the genes utilizing non-standard start codons were filtered. The RSCU plot was created with the online tool Genepioneer (http://112.86.217.82:9929 (accessed on 6 January 2025)). An RSCU value greater than 1 indicates a higher frequency of codon usage, while a value less than 1 indicates a lower frequency [63].
4.4. Phylogenetic Analysis
The performance of phylogenetic reconstruction was tested based on the 19 newly generated sequences combined with an additional 12 Saxifraga species downloaded from NCBI, using Ribes fasciculatum Siebold & Zucc. var. chinense Maxim. (MH191388) and Itea chinensis Hook. f. & Arn. (NC_037884) as the outgroups. The 12 downloaded Saxifraga species include (i) S. cinerascens Engl. & Irmsch. (NC_070452), S. nangxianensis J. T. Pan (NC_070492), S. filicaulis Wall. ex Ser. (NC_070461), S. hemisphaerica Hook. f. & Thoms. (NC_070471), S. tsangchanensis Franch. (NC_070517), S. umbellulata Hook. f. & Thoms. (NC_070518) and S. brevicaulis Harry Sm. (NC_070447) from sect. Ciliatae; (ii) S. fortunei Hook. f. (NC_070463), S. rufescens Balf. f. (NC_070504) and S. stolonifera Curt. (NC_037882) from sect. Irregulares Haw.; and (iii) S. cernua L. (NC_070450) and S. granulifera Harry Sm. (NC_070468) from sect. Mesogyne Sternb. was downloaded from the NCBI database. In total, 31 Saxifraga species from eight sections, plus two outgroups, were included to conduct phylogenetic analysis. Common protein coding genes (PCGs) were extracted using PhyloSuite and concatenated into a single matrix for each species. The concatenated sequences were then aligned using MAFFT. Phylogenetic relationships were reconstructed by means of maximum likelihood (ML) and Bayesian inference (BI) using IQ-TREE [64] and MrBayes [65], respectively, as implemented in PhyloSuite. The best-fitting models were GTR + F+I + G4 according to BIC and GTR + I + G, separately. Bootstrap support (BS) values and posterior probabilities (PP) were calculated using 1000 replications. Phylogenetic trees were visualized and adjusted with iTOL v.6 [66].
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ebersbach J. Schnitzler J. Favre A. Muellner-Riehl A.N. Evolutionary radiations in the species-rich mountain genus Saxifraga LBMC Evol. Biol.2017171192854538610.1186/s 12862-017-0967-2PMC 5445344 · doi ↗ · pubmed ↗
- 2Webb D.A. Gornall R.J. Saxifrages of Europe Helm London, UK 1989
- 3Pan J.T. Gornall R.J. Ohba H. Saxifraga Flora of China Wu C.Y. Raven P.H. Science Press Beijing, China Missouri Botanical Garden Press St. Louis, MO, USA 2001 Volume 8
- 4Gao Q.B. Li Y.H. Gornall R.J. Zhang Z.X. Zhang F.Q. Xing R. Fu P.C. Wang J.L. Liu H.R. Tian Z.Z. Phylogeny and speciation in Saxifraga sect. Ciliatae (Saxifragaceae): Evidence from psb A-trn H, trn L-F and ITS sequences Taxon 20156470371310.12705/644.3 · doi ↗
- 5Ebersbach J. Muellner-Riehl A.N. Favre A. Paule J. Winterfeld G. Schnitzler J. Driving forces behind evolutionary radiations: Saxifraga section Ciliatae (Saxifragaceae) in the region of the Qinghai-Tibet Plateau Bot. J. Linn. Soc.201818630432010.1093/botlinnean/box 100 · doi ↗
- 6Ebersbach J. Tkach N. Röser M. Favre A. The role of hybridisation in the making of the species-rich arctic-alpine genus Saxifraga (Saxifragaceae)Diversity 20201244010.3390/d 12110440 · doi ↗
- 7Liu L. Xu X. Zhang L. Li Y. Shrestha N. Neves D.M. Wang Q. Chang H. Su X. Liu Y. Global patterns of species richness of the Holarctic alpine herb Saxifraga: The role of temperature and habitat heterogeneity J. Plant. Ecol.20221523725210.1093/jpe/rtab 085 · doi ↗
- 8Soltis D. Soltis P. Endress P. Chase M. Manchester S. Judd W. Majure L. Mavrodiev E. Phylogeny and Evolution of the Angiosperm: Revised and Updated Edition The University of Chicago Press Chicago, IL, USA 2018