Exploring lncRNA-Mediated Mechanisms in Muscle Regulation and Their Implications for Duchenne Muscular Dystrophy
Abdolvahab Ebrahimpour Gorji, Zahra Roudbari, Kasra Ahmadian, Vahid Razban, Masoud Shirali, Karim Hasanpur, Tomasz Sadkowski

TL;DR
This paper explores how long non-coding RNAs influence muscle regulation and their role in Duchenne Muscular Dystrophy.
Contribution
The study provides a synthesis of lncRNA roles in myogenesis and their implications for DMD progression.
Findings
LncRNAs regulate skeletal myogenesis and are linked to muscular diseases like DMD.
Some lncRNAs control genes or miRNAs, affecting muscle cell function and DMD development.
The research enhances understanding of lncRNA regulatory functions in muscle growth and DMD.
Abstract
Duchenne muscular dystrophy (DMD) manifests as a hereditary condition that diminishes muscular strength through the progressive degeneration of structural muscle tissue, which is brought about by deficiencies in the dystrophin protein required for the integrity of muscle cells. DMD is among four different types of dystrophinopathy disorders. Current studies have established that long non-coding RNAs (lncRNAs) play a significant role in determining the trajectory and overall prognosis of chronic musculoskeletal conditions. LncRNAs are different in terms of their lengths, production mechanisms, and operational modes, but they do not produce proteins, as their primary activity is the regulation of gene expression. This research synthesizes current literature on the role of lncRNAs in the regulation of myogenesis with a specific focus on certain lncRNAs leading to DMD increments or…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
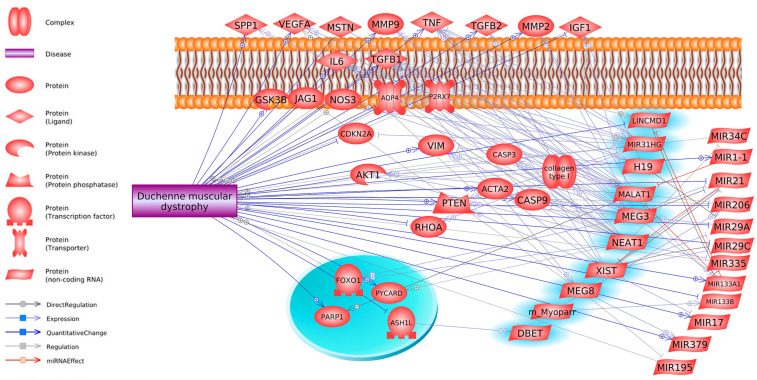 Figure 1
Figure 1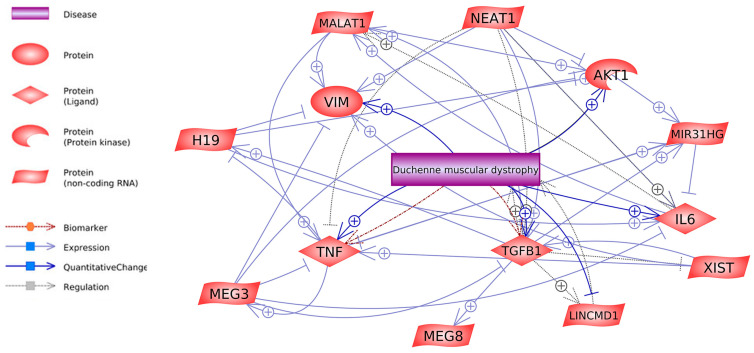 Figure 2
Figure 2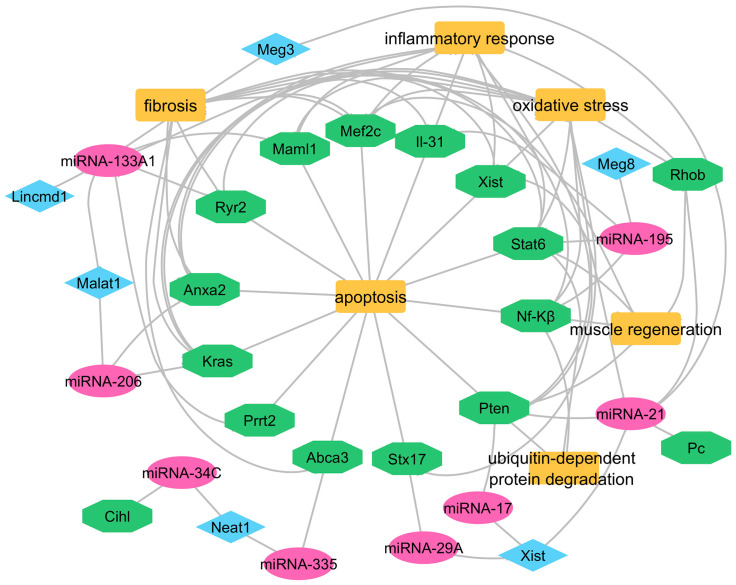 Figure 3
Figure 3| LncRNA | Chr Location | Effect on Genes | Genes | Chr Location | References | DMD Effect on Gene | References |
|---|---|---|---|---|---|---|---|
| Meg3 | 14 | positive |
| 12 | [ | positive | [ |
| Meg3 | 14 | negative |
| 14 | [ | positive | [ |
| Meg3 | 14 | negative |
| 7 | [ | positive | [ |
| Meg3 | 14 | negative |
| 20 | [ | positive | [ |
| Meg3 | 14 | negative |
| 19 | [ | positive | [ |
| Meg3 | 14 | negative |
| 6 | [ | positive | [ |
| Meg3 | 14 | positive |
| 4 | [ | - | - |
| Meg3 | 14 | positive |
| 1 | [ | positive | [ |
| Meg3 | 14 | positive |
| 13 | [ | - | - |
| Meg3 | 14 | positive |
| 16 | [ | positive | [ |
| Meg3 | 14 | positive |
| 10 | [ | positive | [ |
| Neat1 | 11 | negative |
| 14 | [ | positive | [ |
| Neat1 | 11 | negative |
| 1 | [ | positive | [ |
| Neat1 | 11 | positive |
| 10 | [ | positive | [ |
| Neat1 | 11 | positive |
| 4 | [ | - | - |
| Neat1 | 11 | positive |
| 13 | [ | - | - |
| Neat1 | 11 | positive |
| 7 | [ | positive | [ |
| Neat1 | 11 | positive |
| 16 | [ | - | - |
| Neat1 | 11 | positive |
| 19 | [ | positive | [ |
| Xist | X | positive |
| 10 | [ | positive | [ |
| Xist | X | positive |
| 19 | [ | positive | [ |
| Xist | X | negative |
| 19 | [ | positive | [ |
| Malat1 | 11 | positive |
| 18 | [ | negative | [ |
| Malat1 | 11 | negative |
| 16 | [ | positive | [ |
| Malat1 | 11 | positive |
| 14 | [ | positive | [ |
| Malat1 | 11 | positive |
| 20 | [ | positive | [ |
| Malat1 | 11 | positive |
| 7 | [ | positive | [ |
| Malat1 | 11 | positive |
| 1 | [ | positive | [ |
| Lnc31 | 9 | negative |
| 3 | [ | positive | [ |
| Lnc31 | 9 | positive |
| 10 | [ | positive | [ |
| H19 | 11 | positive |
| 12 | [ | positive | [ |
| H19 | 11 | positive |
| 14 | [ | positive | [ |
| H19 | 11 | positive |
| 7 | [ | positive | [ |
| H19 | 11 | positive |
| 6 | [ | positive | [ |
| Meg8 | 14 | positive |
| 20 | [ | negative | [ |
| Meg8 | 14 | positive |
| 6 | [ | positive | [ |
| Dbet | 4 | positive |
| 1 | [ | negative | [ |
- —Science development fund of the Warsaw University of Life Sciences—SGGW
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer-related molecular mechanisms research · RNA modifications and cancer · RNA Research and Splicing
1. Introduction
Duchenne muscular dystrophy (DMD) is an X-linked recessive muscular dystrophy caused by pathogenic variants in the dystrophin gene (DMD), affecting patients from early childhood [1]. The majority of DMD patients aged 2–3 years old have muscle breakdown that leads to progressive weakness, more prominent initially in the proximal lower limbs, that rapidly leads to wheelchair dependence by early adolescence, as described in the natural history of the disease [2,3]. Deaths among DMD patients occur between 20 and 40 years because their muscles weaken to extreme debilitation. Thus, the primary cause of death is end-stage cardiomyopathy, often complicated by superimposed respiratory infections or acute episodes [4]. Various studies and sources have different reports concerning DMD prevalence as well as its incidence at birth. Current research suggests DMD affects 0.9 to 16.8 of 100,000 males, but birth prevalence studies suggest DMD occurs 1.5 to 28.2 times in 100,000 live-born male children. The numerous scientific estimates reflect the difficulty in estimating DMD incidence in males [5].
The pathogenetic variants of the DMD gene cause progressive deterioration of the skeletal muscle, the interplay of damage resulting from disrupted muscle fibers, and severe cycles of degeneration and regeneration, leading to progressive muscle atrophy and replacement with fibrotic and adipose tissues during the disease process [6]. The myogenesis of skeletal muscle is a multi-step process with various discrete stages characterized by the co-existence of myogenic regulatory factors (MRFs), which include Myf5, myogenic differentiation antigen (MyoD), myogenin (MyoG), and Mrf-4, as well as myosin heavy chain (Mhc) [7]. From a pathogenetic perspective, dystrophin deficiency leads to progressive muscle deterioration, inflammation, and pro-oxidative/mitochondrial stress [8,9]
LncRNA has now been acknowledged as a crucial factor directly interacting with multiple myogenic regulatory factors. The long non-coding RNA class comprises non-coding RNA species that exceed 200 nucleotides in length [4,10,11]. These RNA species play imperative roles by modulating gene expression at transcriptional and post-transcriptional levels [12,13]. LncRNAs differ from mRNAs as they do not have an open reading frame (ORF), having around 2.8 exons, whereas proprotein-coding RNA has 11 exons and low abundance [14]. LncRNAs can indirectly regulate gene expression in three ways: direct activation of transcription factors, miRNA sponge, enzymatic recruitment to genomic loci, and sequence changes [15,16]. Research has shown that some non-coding RNAs control muscle growth and development, albeit their genetic analysis boundaries are poorly understood [17,18].
Due to their underuse in genetic analysis, these RNAs have not received much focus in genetic research on muscle development. The Pathways Studio Software Web Mammalian (V. 12.5.0.2, 2022) was used to filter out matching lncRNAs that potentially influence the development of DMD and their miRNA binding and genetic motifs, and available literature on DMD and lncRNA, for the current review. LncRNA is encoded by more than 56,946 genes (127,802 transcripts) but has little or no potential to produce protein [19]. In addition, lncRNAs are transcribed into some peptides with regulatory functions [20].
Based on high-throughput technologies and bioinformatics analyses, thousands of lncRNAs have been discovered in skeletal muscles, but only a few have been identified as functionally regulated (Table 1). Hou et al. examined the skeletal muscle transcriptome, detecting 322 lncRNAs. The identified lncRNAs regulate the gene function of Csrp3, Myf6, Igfbp, Dcn, Map2k1, Spp1, and Acsl1, which control skeletal muscle development and fatty acid metabolism [19]. The groups of lncRNAs bind proteins to affect the development and deterioration of muscles. Metabolism-related lncRNAs: Gm15441 controls insulin signal transmission while sustaining stable blood glucose levels in skeletal muscles. Pparα activation level rises during fasting periods, leading to Gm15441 gene expression elevation through Pparα-binding sites in its promoter [21]. The protein Gm15441 forms a complex with Txnip and decreases protein amounts, which results in reduced hepatic glucose production [22]. A Spearman correlation analysis showed 3110045C21Rik and Ddr2 expression occur together in type 2 diabetes mellitus (T2DM) patients, which indicates 3110045C21Rik regulates insulin-signaling mechanisms [23]. Fibrosis-related lncRNAs: Furthermore, the overexpression of 3110045C21Rik promotes the up-regulation of E-cadherin (Cdh1) while suppressing the expression of alpha-smooth muscle actin (Acta2) and Transforming Growth Factor Beta 1 (Tgfb1), key markers associated with the development and progression of fibrosis [24]. Myogenesis and Apoptosis-related lncRNAs: Recent literature identifies several lncRNAs related to skeletal muscle atrophy, including SYISL, Mir22hg, Myoparr, Pvt1, RP11-253E3.3, PRKG1-AS1, lncDLEU2, Chronos, lnc-ORA, lncMUMA, Atrolnc-1, H19, HOTAIR, Malat1, PVT1, Gm15441, 3110045C21Rik, Gm20743, LncEDCH1, ZFP36L2-AS, LncIRS1, SMUL, lnc-mg, and MYH1G-AS. These lncRNAs regulate key processes in muscle atrophy through diverse mechanisms, including modulation of protein synthesis, ubiquitin-proteasome-mediated protein degradation, and myogenic differentiation [25]. A Weighted Gene Co-expression Network Analysis (WGCNA) of goat skeletal muscle in developmental stages revealed that LNC_011371 (targets 74 genes, including Mb and Clic5, that are up-regulated after birth), LNC_007561 (targets Tcf4 that regulates myogenesis), and LNC_001728 (binds to S100A4 gene and promotes cardiomyocyte production and increases myocardial cell number by inhibiting apoptosis) involve in muscle structure formation, p53-signaling pathways, and MAPK-signaling pathways [26].
2. LncRNAs in Muscle Development
The myogenic differentiation process benefits from Ppp1r1b-lncRNA positive regulatory action. The Ppp1r1b and polycomb repressive complex 2 (PRC2) interaction stimulates the transcription of myogenic transcription factors in a mouse C2C12 myoblast cell line and human skeletal myoblasts. The absence of Ppp1r1b leads Ezh2 to enhance its binding activities on the regulatory regions of these myogenic transcription factors. The suppression of MyoD, Myogenin, and Tbx5, along with sarcomere proteins, becomes very evident. The research indicates that Ppp1r1b functions as a regulatory factor during the early stages of muscle development and heart formation [27]. One of the essential lncRNAs that regulates some transcription factors and miRNAs is the myocardial infarction-associated transcript (Miat). This lncRNA can indirectly regulate some biological processes in muscle, such as cell proliferation, cell differentiation, regeneration, and myogenesis. For instance, Miat upregulates the expression of the transcription factor Foxo1 by sponging miR-139-5p [38], and this gene regulates myogenic differentiation and growth, muscle atrophy, and glycemic properties [39,40]. Moreover, Miat indirectly regulates the expression of Zeb1 by acting as a sponge and repressing miR-150. Expression of Zeb1 has been reported in numerous tissues, including the immune system, and significantly regulates muscle and lymphoid differentiation [41]. Notably, both Zeb1 and Zeb2 are pivotal elements that modulate the TGF-β-mediated signaling pathway through their interaction with Smad proteins to facilitate the recruitment of co-activators or co-repressors [42]. Current studies demonstrate that Miat operates directly to control three transcription factors named Dnmt1, Dnmt3a, and Dnmt3b. Research findings indicate that decreasing Miat levels limits attachment to these transcription factors [28]. Phytohemagglutinin stimulation increased apoptosis and inhibited cell proliferation in response to suppression of the Dnmt1 protein. Evidence shows that the gene modifier exerts substantial control over essential biological operations within immune cells [43]. A feedback loop between Dnmt3b and miR-125b controls vascular smooth muscle cell responses to homocysteine exposure [44]. Studies demonstrate that MyoG attaches to the promoter of this lncRNA to cause its expression activation. It was also shown that enhanced lncRNA-1700113A16RIK expression accelerates the differentiation process of MuSC cells, while decreased expression adversely affects the function of cells. Moreover, this lncRNA directly interacts with the 3′UTR of myogenic genes, including the myogenic transcription factor Mef2d, subsequently enhancing its translation [29].
Investigations into lncRNA expression patterns during skeletal muscle development identified ten lncRNAs, which displayed maximal correlations with known protein-coding genes. Hub lncRNAs consisting of lnc-22988, lnc-372289, and lnc-482286 exhibited positive correlation while connecting with 156 protein-coding genes, including the crucial skeletal muscle developmental genes Acta1, Eno3, Myl1, Myom1, Myoz1, Neb, Ryr1, and Tnnc2. The three lncRNAs are linked to various genetic elements that potentially control muscle proliferation and differentiation and global developmental pathways [30]. The expression of miR-675-3p and miR-675-5p increases due to H19 regulation in skeletal muscle cells, and these miRNAs reduce Smad1, Smad5, and Cdc6 gene activity to drive both skeletal muscle differentiation and regeneration [31]. Moreover, an increase in H19 results in the higher expression of dual-specificity phosphatase 27 (Dusp27) at the posttranscriptional level, promotes AMPK pathway activity in muscle cells, and stimulates glucose uptake and mitochondrial biogenesis. The decline of this lncRNA expression in human and mouse muscles leads to muscle insulin resistance, while high concentrations of H19 can enhance muscle insulin sensitivity [32]. In the embryonic stage, lncRNAs such as H19, Alien, and Miat are identified to have a key regulatory function. However, in cardiac development, their role is unclear, and H19 displays a dynamic epicardial, myocardial, and endocardial expression during cardiac development [45].
Several lines of evidence demonstrate that Chronos plays a pivotal role in hypertrophy. Using inducible Akt1 transgenic mice to investigate age-related lncRNAs revealed that Chronos inhibits Akt1 and hypertrophic growth. Studies in vivo and in vitro show that Chronos inhibition’s effect on the Bmp-signaling pathway in general [46] and the Bmp7 ligand specifically [47], leading to increased myofiber hypertrophy [48]. Furthermore, the Bmp gene is essential for developing blood vessels required for steady muscle states [49].
3. LncRNA and miRNA—Working Together in Muscles
Skeletal muscle formation depends on lncRNAs and miRNAs, which operate within a complex regulatory system to control myogenesis. They regulate chromatin, activate transcription, act as miRNAs sponges, modulate RNA stability and translation, and, in some cases, encode micropeptides. MiRNAs regulate myogenic gene expression precisely, while lncRNAs, often functioning as sponges, modulate miRNA activity to fine-tune proliferation, differentiation, and regeneration. This coordinated regulation is critical for muscle development and holds promise for diagnosing and treating muscle diseases, such as muscular dystrophies [50]. A direct relationship exists between lncRNA and miRNA expression patterns, combining regulatory mechanisms across biological operations. The lncMgpf functions as an example by influencing the expression level of miR-135a-5p. Through this interaction, lncMgpf weakens the inhibitory effects of miR-135a-5p, consequently elevating the expression of Mef2c. This action further increases human antigen R (HuR)-mediated mRNA and regulates muscle development genes like MyoD and MyoG [51]. Another study shows that AK003290 in mice and its homologous lncRNA AK394747 in pigs and MT510647 in humans positively regulate MyoD expression and enhance myogenic differentiation of muscle cells [5]. During muscle differentiation in C2C12 cells, miR-487b regulates Wnt5a, a key player in myogenesis, and promotes muscle differentiation and regeneration. A study on lncRNA shows that muscle anabolic regulator 1 (MAR1) may regulate miR-487b and positively correlate with muscle differentiation. In addition, this lncRNA significantly enhanced mRNA and protein levels of myogenic markers (MyoD, MyoG, Mef2c, and Myf5), with highly expressed formation of myotubes in mouse skeletal muscle [33]. Moreover, overexpression of the intronic sense-overlapping lncRNA known as Syisl adversely affects the differentiation process of C2C12 cells. Elevated Syisl expression delays cellular differentiation while promoting cell proliferation [52]. Also, Syisl homologs in humans (designated hSyisl) and pigs (designated pSyisl) regulate myogenesis through interactions with Ezh2 and regulate muscle atrophy and sarcopenia [34].
3.1. Cell Cycle and Differentiation Regulators
Specific lncRNAs function during the regulation of multiple myogenesis steps, and their depletion or dysregulation impairs cellular capacity to achieve cell cycle exit, resulting in aberrant proliferation. LncMyoD (Gm45923) is a regulatory lncRNA that binds directly to Igf2-mRNA-binding protein 2 (IMP2) during myoblast differentiation, downregulating the IMP2-mediated translation of proliferation-associated genes like N-Ras and c-Myc [53]. Meanwhile, miR-370-3p, which is directly targeted by lncMyoD, promotes myoblast proliferation and hinders myogenic differentiation of the C2C12 cell line [35]. Another lncRNA, Dum, follows the path of myoblast differentiation when stimulated by MyoD. Silencing Dppa2 and suppressing the MyoD–Dum–Dppa2 complex formation further augments myoblast differentiation and damage-induced muscle regeneration. A Dum knockdown could affect satellite cell activation, proliferation, or self-renewal capacity [36]. Furthermore, a network analysis of lncRNA-miRNA-gene interactions related to myogenesis led to the identification of lncIrs1, which regulates myoblast proliferation and differentiation [54]. LncRNA-Irs1 influences Irs1 expression downstream of the Igf1-receptor-signaling pathway. Elevating lncIrs1 expression in breast muscle mitigates muscle atrophy and enhances muscle weight. Its overexpression boosts the phosphorylation of AKT within the IGF-1 pathway [52].
3.2. Atrophy Regulators
Research on lncRNA FKBP1C has shown that it is a developmental regulator of skeletal muscle tissue that prevents myoblast replication while supporting differentiation, mostly in fast and slow muscle fibers. Research on the Myh1b gene silencing primarily showed effects in fast muscle fibers. The lncRNA enhances Myh1b expression through cis-regulation, promoting protein stability, and influencing myoblast development [37]. The co-expression network analysis highlighted three important lncRNAs linked to skeletal muscle and atrophy, which are Ac004797.1, Prkg1-As1, and Grpc5d-As1. Research results demonstrated that the knockdown of Prkg1-As1 increased MyoD, MyoG, and Mef2c gene expression while reducing apoptosis in myoblast cells and the mortality rate [55].
The decoy activity of lncRNAs results in miRNA sequestration, which blocks their interaction with target mRNAs and disrupts miRNA-mRNA communication networks [47,56]. MiR-127 interferes with the retrotransposon-like one protein (Rtl1) sense transcript, reducing Rtl1 protein production. The protein is essential throughout muscle regeneration and present within regenerating and dystrophic muscle tissue [57]. Another research, by Liu et al. (2020), demonstrates that miR-324 limits C2C12 myoblast differentiation while accelerating intramuscular lipid accumulation through regulation by the lncRNAs Dum and Pm20d1 [58]. Moreover, it was shown that lncRNA-Miat functions as a direct target of miR-214, demonstrating that inhibition of this miRNA restores hepatocellular carcinoma cell proliferation and invasion by counteracting the suppression of Miat expression levels [59].
4. LncRNA in DMD Disease
Atypical expression of lncRNAs is linked to diverse muscular disorders, most notably DMD, as outlined in Table 2. Current investigations have revealed that lncRNAs can regulate gene expression and translation, significantly influencing the progression of pathological states during cellular and muscular differentiation and development. Alterations in the expression of some lncRNAs are evident in the skeletal muscles of DMD patients (Figure 1).
Bovolenta et al. (2012) extensively investigated the DMD gene to elucidate how lncRNAs regulate gene expression and how nuclear lncRNAs modulate promoter regions to control muscle-specific isoform expression. Most regulatory lncRNA elements are found in intronic regions and nuclear spaces [60]. A detailed study based on bioinformatics identified Xist, Al132709, Linc00310, and Aldh1l1-As2 as key lncRNA regulators of DMD secondary processes, such as muscle degeneration and fibrosis. The diverse biological functions of these lncRNAs entail tumor-promoting capacity and ventricular septum development [61]. Dystrophin stabilizes the sarcolemma and is a scaffold for various intracellular signaling pathways, including nitric oxide synthase (NOS) signaling and the PI3K/Akt pathway [62]. LncRNAs, such as H19 and Malat1, are implicated in modulating these pathways. For instance, H19 can promote Akt1 expression, potentially counteracting muscle atrophy in dystrophic muscles, while lnc-31 influences myoblast differentiation by regulating myogenic gene expression [60,63]. These lncRNAs may act as upstream regulators or downstream effectors of dystrophin-dependent signaling, indicating a complex interplay that could amplify or mitigate the signaling defects caused by dystrophin deficiency [64].
For DMD research, a continued examination of lncRNAs occurred in the C2C12 cell line and human myoblasts. The study showed that lnc-31 exists at higher levels in DMD patient subjects than in individuals without DMD. The nuclear-based precursor molecule that produces miR-31 also creates the significant factor lnc-31, which guides the differentiation of precursor myoblasts. When lnc-31 is absent, the elevated expression of myogenin along with Atp2a1 causes rapid myogenic differentiation, which becomes difficult to reverse. This implies that lnc-31 is critical in controlling the transition from cell-cycle exit to terminal differentiation [119]. Another notable lncRNA in skeletal muscle regeneration and dystrophic muscles resides in the gene’s intron 44 (lncRNA44s2). This lncRNA exhibits expression during myogenesis in primary human myoblasts. It demonstrates activity linked to myogenesis, indicating its possible role in muscle differentiation and its potential as a disease-progression biomarker [63].
It is interesting to note that specific lncRNAs impact the progression of skeletal muscle atrophy. Physiological studies on different muscle atrophy models show depressed levels of lncMaat. Restoring or increasing lncMaat expression might then counteract multiple forms of atrophy. LncMaat inhibits miR-29b transcription through Sox6 and simultaneously represses Mbnl1 expression from the same regulatory module, leading to muscle atrophy regardless of miR-29b activity. Mbnl1 overexpression shows excellent potential for preventing muscle atrophy [120]. The literature data show that Long Intergenic Non-Protein Coding RNA, Muscle Differentiation 1 (LincMD1), becomes vital in understanding the development of DMD, as its expression is significantly reduced in myoblasts from DMD patients compared to healthy controls. The reduced levels of LincMD1 are connected to the delayed expression and development of Mhc and myogenin muscle-specific markers in DMD tissue. The therapeutic potential of LincMD1 is demonstrated by its ability to enhance Myog and Mef2c expression in DMD patient-derived myoblasts, promoting myogenic differentiation [121].
Scientific evidence reveals a significant relationship between serine/threonine-protein kinase MRCK alpha (Mrckα) and α-synuclein (Snca) with lncRNA-H19. When Mrckα and Snca actively bind, they displace H19 from interacting with DMD gene segments to promote better myotube development and more efficient cell fusion in skeletal muscle cells. The administration of AGR-H19-gain-of-function enhances WT mouse muscle tissue development, metabolic performance, and muscle mass increase [120]. The lncRNA H19 accelerates muscle regeneration by activating the miR-675-3p and miR-675-5p, which regulate myogenic pathways [31]. The influence of specific non-muscle-specific lncRNAs shapes muscle proliferation and differentiation processes and contributes to both normal and pathological conditions. The cellular proliferation inhibitor named maternally expressed gene 3 (Meg3) functions to regulate muscle metabolism, along with glucose tolerance functions. Limited lncRNA Meg3 activity impairs glucose tolerance and negatively affects muscle metabolism [122,123]. According to Butchart et al. (2018), the expression of the Meg3 gene remained the same between the atrophy disease model, mdx skeletal muscles, and normal C57 mouse muscle tissues [124].
Many lncRNAs work together to control Tnf gene activity, which might become a diagnostic marker for DMD [125]. Studies demonstrate elevated Tnf gene expression levels in DMD patients compared to healthy samples [9,70]. This regulation involves a spectrum of lncRNAs, including Meg3, Xist, lnc-31 (Mir31hg), Neat1, and Malat1, culminating in intricate control over this key gene’s activity (see Figure 2).
A study showed that Meg3 overexpression decreased the pro-inflammatory cytokines Tnf-α, Il-6, and Il-1β levels [126]. The opposite results occur after silencing of Meg3 [127,128]. The increased expression level of Meg3 leads to diminished cell multiplication and reduced T-bet, Ifn-β, and Tnf-α levels. The reduction in these factors results in substantial increases in cell multiplication. The functions of lnc-31 and Neat1 in Tnf-α expression regulation match each other. The inhibitory action of lnc-31 on Tnf-α expression facilitates cell growth that might help reduce hypoxia-generated cell impairment. Neat1 exhibits anti-inflammatory properties that provide potential benefits against DMD inflammation when combined with associated treatments [129].
The expression profile of Tnf-α can be changed by several lncRNAs, such as Malat1 and Xist, because they activate the gene for Tnf-α production. Lowering of lncRNA Malat1 yields two primary effects on skeletal muscle cells. It leads to decreased Il-6, Il-8, Tnf-α serum levels, triggers cell death, and causes Akt-1 protein activation [130]. The reduction in Xist expression inhibits Tnf-α protein production and stops the Tnf-α/Rankl-signaling pathway activation process. The expression levels of Tnf-α significantly increase when Xist is overexpressed. Elevated levels of Xist following bone fractures hinder cell proliferation and differentiation, whereas reducing Xist expression promotes cell growth and regeneration [131]. The literature demonstrates that this lncRNA helps activate Pten by blocking the action of miR-17. Research has revealed that Xist silencing results in elevated levels of miR-17 in patients who experience type A aortic dissection [132]. The study of Yue et al. 2021 reported that Pten expression levels increased within the DMD and mdx mouse muscles, showing signs of muscular dystrophy. Reports indicate that Xist lncRNA indirectly supports the progression of DMD [84].
The clinical severity of DMD depends strongly on the functioning of the Tgfb1 gene. Evaluation of Tgfb1 expression enhances fibroblast multiplication and collagen synthesis while transforming fibroblasts into myofibroblasts [133]. The lncRNA molecules Neat1, Xist, and Meg3 maintain control over this gene. In molecular studies, in vitro experiments have shown that Xist can stimulate Tgfb1 expression by upregulating miR-185 [134]. Neat1 stimulates Tgfb1 overexpression through a competitive sponge action against miR-339-5p [135]. The RNA analysis demonstrates that Meg3 controls the cellular transition known as Epithelial-to-Mesenchymal Transition (EMT) through its ability to suppress Tgfβ signaling. Interestingly, inhibiting Tgfβr1 or its downstream effectors, like RhoA, p38 MAPK, or Snai2, was proven to successfully reinstate facets of myogenic fusion and differentiation in vitro. The Meg3 functions as an inhibitor of Tgfbi or Tgfb1 expression while integrating a modification of anti-myogenic EMT-promoting factors Tgfβ, RhoA, and Snai2 in myoblasts and injured skeletal muscle [136] (Figure 2). The expression of Akt1 protein is upregulated through the regulatory actions of lncRNAs H19 and Malat1, which facilitate muscle hypertrophy in mdx mice [68].
5. How lncRNAs Can Control DMD Through miRNA and Gene Networks
Recent bioinformatics studies revealed distinct mechanisms by which Meg3 functions in its biological processes. The study shows that Meg3 functions as a miR-21-5p sponge, suggesting that this miRNA targets the 3′ untranslated region of the Pten sequence. Due to the overexpression of Meg3, Pten gene expression increases, leading to control of Pi3k/Akt signaling [137]. A third lncRNA, termed Neat1, reduces the expression levels of Akt1. The overexpressed miR-214 subsequently leads to elevated Pi3k, Akt, P-Akt, and Vegf level. The activating effect is achieved through Pten reduction. The expression of this specific lncRNA opposes the protective role of miR-214 on cerebral ischemia–reperfusion damage. By restoring Pten levels, the inhibition of Pi3k together with Akt, P-Akt, and Vegf production occurs [138].
Research indicates that serum interleukin-6 (IL-6) concentration is typically elevated in patients with DMD who are steroid-naïve or untreated compared to those treated with glucocorticoids. [139,140]. Different cell regulators, lncRNAs such as Neat1, H19, Meg3, and lnc-31, maintain control over this gene. The expression of Il-6 and Il-8 benefits from increased levels of lncRNA Neat1. This regulation is achieved by downregulating the expression of miR-181c [59], miR-139 [141], and miR-144-3p [142]. Similarly, H19 contributes to the regulation of IL-6 in a comparable manner. It promotes the expression of this gene by acting as a sponge for miR-let-7a, contributing to an increase in vascular inflammation. In contrast, Meg3 and lnc-31 exert inhibitory effects on the secretion of Tnf-α and Il-6 [127,143]. Vim represents another gene under the influence of specific lncRNAs. Some studies reveal notably elevated Vim gene expression in DMD and dystrophic hearts [144,145], but its exact function is still unclear. Xist lncRNA emerges as a potential regulator, possibly inhibiting miR-92b and freeing miR-92b from the 3′ UTR of Smad7. This process potentially triggers the activation and subsequent upregulation of Vim expression [146]. Additionally, Zhang et al. found that miR-143 and Malat1 directly regulate the Vim and epithelial–cadherin protein levels, but lower miR-143 expression combined with elevated Malat1 [147]. Conversely, higher Neat1 levels lead to an elevation of E-cadherin, Neural–cadherin protein, and Vim protein simultaneously [87]. Instead, the lncRNA H19 acts as a regulator that affects the expression of Vim, Zeb1, and Zeb2 by competing with endogenous RNAs, specifically miR-138 and miR-200a [148]. Furthermore, the overexpression of Meg3 leads to downregulation of Vim and fibronectin mesenchymal markers as it inhibits gastric cancer cell proliferation [149].
Interactions between lncRNAs and miRNAs are highly significant, as they play a crucial role in regulating gene expression. The expression of muscle-specific miRNAs regulates the modulation of muscle differentiation and homeostasis, while their patterns show changes in conditions like myocardial infarction and DMD, together with other myopathies [150,151] (Table 3, Figure 3). Cell migration and invasion are inhibited through the interaction of miRNA that simultaneously affects the gene expression of Anxa2 and Kras. The removal of miR-206 leads to worse and more rapid symptoms in mouse models of DMD [152]. Mice expressing the mature form of this miRNA demonstrate enhanced muscle tissue repair and a slower progression of Duchenne muscular dystrophy [153]. Also, the overexpression of miR-1 leads to a decrease in Malat1 expression [154]. Interestingly, miR-1 has demonstrated its potential to suppress breast cancer development by downregulating Kras and Malat1 transcription [155]. Investigations into miR-1 levels have shown significant elevation in DMD patients compared to healthy control subjects [156,157].
6. Conclusions
Recently, the importance of lncRNAs has increased because they can regulate many genes and are master regulators of various genetic, epigenetic, and biological processes essential to developing multiple disorders but are also potent regulators of muscle regeneration, affecting genes involved in DMD disease. The mechanisms of lncRNAs-mediated gene regulation discussed in this manuscript clarify that lncRNAs can regulate DMD-related gene expression at the post-transcriptional level, with a less abundant but significant role in post-translational regulation. Post-transcriptionally, lncRNAs such as Miat, H19, Meg3, and lnc-31 modulate mRNA stability, splicing, translation, and miRNA interactions (e.g., miRNA sponging, direct mRNA binding), influencing myogenic differentiation and DMD progression. Post-translationally, lncRNAs like H19 and FKBP1C regulate protein stability and signaling pathways (e.g., Akt, AMPK), highlighting their critical role in skeletal muscle development and DMD pathology. Adeno-associated virus (AAV)-mediated delivery holds promise for DMD therapy, supported by precedents in other diseases where AAV vectors delivered lncRNAs like H19 (cancer), Malat1 (vascular disease), and Neat1 (neurological disorders) to achieve therapeutic effects.
However, while the presented examples suggest potential roles for lncRNAs in DMD pathogenesis and therapeutic applications, further studies are required to establish causal relationships and validate their therapeutic potential.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ousterout D.G. Kabadi A.M. Thakore P.I. Majoros W.H. Reddy T.E. Gersbach C.A. Multiplex CRISPR/Cas 9-Based Genome Editing for Correction of Dystrophin Mutations That Cause Duchenne Muscular Dystrophy Nat. Commun.20156624410.1038/ncomms 724425692716 PMC 4335351 · doi ↗ · pubmed ↗
- 2Duan Y. Song B. Zheng C. Zhong Y. Guo Q. Zheng J. Yin Y. Li J. Li F. Dietary Beta-Hydroxy Beta-Methyl Butyrate Supplementation Alleviates Liver Injury in Lipopolysaccharide-Challenged Piglets Oxidative Med. Cell. Longev.20212021 e 554684310.1155/2021/5546843 PMC 803502233868570 · doi ↗ · pubmed ↗
- 3Mercuri E. Bönnemann C.G. Muntoni F. Muscular Dystrophies Lancet 20193942025203810.1016/S 0140-6736(19)32910-131789220 · doi ↗ · pubmed ↗
- 4Cai A. Kong X. Development of CRISPR-Mediated Systems in the Study of Duchenne Muscular Dystrophy Hum. Gene Ther. Methods 201930718010.1089/hgtb.2018.18731062609 · doi ↗ · pubmed ↗
- 5Crisafulli S. Sultana J. Fontana A. Salvo F. Messina S. TrifiròG. Global Epidemiology of Duchenne Muscular Dystrophy: An Updated Systematic Review and Meta-Analysis Orphanet J. Rare Dis.20201514110.1186/s 13023-020-01430-832503598 PMC 7275323 · doi ↗ · pubmed ↗
- 6Dowling J.J. Weihl C.C. Spencer M.J. Molecular and Cellular Basis of Genetically Inherited Skeletal Muscle Disorders Nat. Rev. Mol. Cell Biol.20212271373210.1038/s 41580-021-00389-z 34257452 PMC 9686310 · doi ↗ · pubmed ↗
- 7Tajbakhsh S. Skeletal Muscle Stem Cells in Developmental versus Regenerative Myogenesis J. Intern. Med.200926637238910.1111/j.1365-2796.2009.02158.x 19765181 · doi ↗ · pubmed ↗
- 8Kumar S. Williams D. Sur S. Wang J.-Y. Jo H. Role of Flow-Sensitive micro RN As and Long Noncoding RN As in Vascular Dysfunction and Atherosclerosis Vasc. Pharmacol.2019114769210.1016/j.vph.2018.10.001PMC 690542830300747 · doi ↗ · pubmed ↗