Cellular Lyso-Gb3 Is a Biomarker for Mucolipidosis II
Seigo Terawaki, Hiroki Nakanishi, Toko Shibuya, Norio Sakai, Takanobu Otomo

TL;DR
Researchers found that a substance called Lyso-Gb3 can serve as a biomarker for mucolipidosis II/III, a rare genetic disorder, and its levels decrease with treatment.
Contribution
Lyso-Gb3 is identified as a novel biomarker for mucolipidosis II/III, which previously lacked reliable biomarkers.
Findings
Lyso-Gb3 levels are elevated in mucolipidosis II/III patient cells compared to normal cells.
Lyso-Gb3 levels decrease after total lysosomal enzyme supplementation in mucolipidosis II/III cells.
Lyso-Gb3 is elevated in knock-out cells of GNPTAB gene models for mucolipidosis II/III.
Abstract
Lysosomal storage diseases are caused by defective lysosomal function, such as impaired lysosomal enzyme activities, which include more than 70 different diseases. Although biomarkers and therapies have been developed to date for some of them, many others remain challenging to diagnose and treat. In this study, an elevated level of Globotriaosylsphingosine (Lyso-Gb3), an already known biomarker for Fabry disease, was confirmed in the knock-out cells of the GLA, GNPTAB, and PSAP genes and models for Fabry, mucolipidosis II/III (ML II/III), and combined saposin deficiency, respectively. Lyso-Gb3 was high in ML II/III patient skin fibroblasts compared with normal cells and was decreased after total lysosomal enzyme supplementation. There have been no useful biomarkers reported in ML II/III until now. This study shows that Lyso-Gb3 is elevated in ML II/III patient cells and is decreased by…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
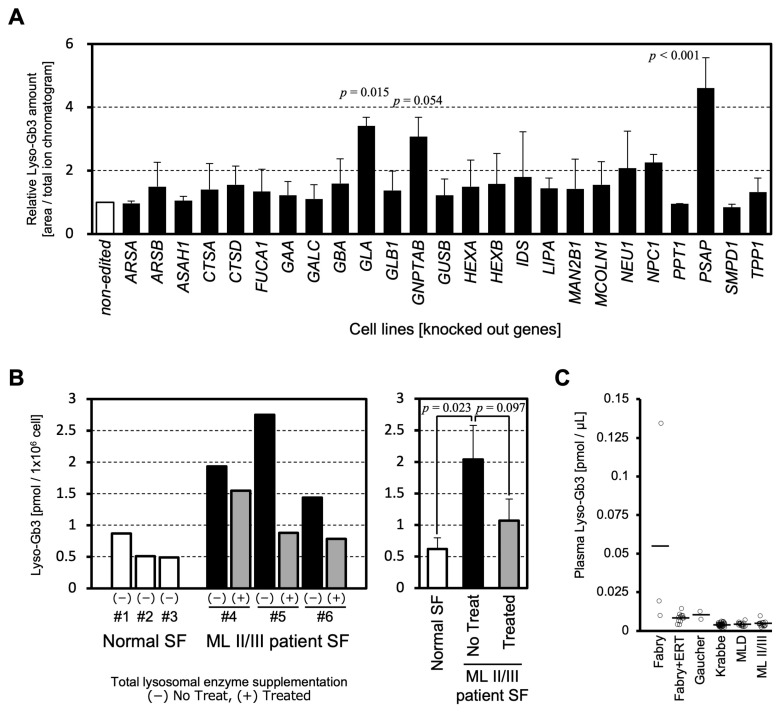 Figure 1
Figure 1- —JSPS KAKENHI
- —AMED
- —Japanese Society for Inherited Metabolic Diseases
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLysosomal Storage Disorders Research · Calcium signaling and nucleotide metabolism · Cellular transport and secretion
1. Introduction
Lysosomal storage diseases (LSDs) are inborn errors of metabolism caused by defective lysosomal functions. LSDs include more than 70 diseases, many of which are caused by lysosomal enzyme deficiency, and the others contain impairments of lysosomal targeting of lysosomal hydrolases, lysosomal transporters, lysosomal environments for degradation, and so on [1].
Lysosomal dysfunction leads to the accumulation of undegraded substrates, resulting in a variety of clinical phenotypes. Deficiency of a lysosomal enzyme leads to the primary accumulation of the enzyme’s substrates. Furthermore, in lysosomes filled with a massive accumulation of undegraded substrates, various hydrolytic enzymatic reactions are thought to be impaired, resulting in secondary accumulation of various substrates due to a general decline in lysosomal function. Indeed, we have reported the disruption of the lysosomal acidic environment in mucolipidosis II/III (ML II/III) and lysosomal acid lipase deficiency (LAL-D) [2,3].
The disruption of intracellular vesicular trafficking is also known to cause LSDs. We reported that a genetic mutation in VPS33A, which is a component of protein complexes for tethering intracellular vesicles, leads to mucopolysaccharidosis-plus syndrome [4]. This novel disease manifests a similar clinical phenotype to LSDs and is speculated to be due to impaired transport of substrates to be degraded by lysosomes [5]. ML II/III is caused by multiple deficiencies of lysosomal enzymes due to the disruption of intracellular trafficking of lysosomal enzymes. In ML II/III, abnormalities in the intracellular transport of lipids and in the distribution of mannose 6-phosphate (M6P) receptors that function in lysosomal enzyme transport are reported [2].
These complex mechanisms of lysosomal dysfunction make it difficult to objectively evaluate the effectiveness of specific treatments for LSDs. Appropriate biomarkers that precisely reflect the severity and progression of diseases, or the effectiveness of therapies, are indispensable for the diagnosis and future development of treatments for LSDs such as ML II/III, in which no valuable biomarkers are available.
In this study, we focused on one of the sphingolipids, Globotriaosylsphingosine (Lyso-Gb3). Lyso-Gb3 is a known biomarker for the diagnosis and disease assessment of Fabry disease, a type of LSD [6,7,8,9]. Fabry disease is caused by a deficiency in the GLA gene that codes for α-galactosidase A, a lysosomal hydrolase. Sphingolipids such as globotriaosylceramide (Gb3) and galactosylceramide are the substrates of α-galactosidase A and accumulate in tissues throughout the body. In particular, Lyso-Gb3, the lyso form of Gb3, has been suggested to be related to the organ damage caused by Fabry disease, such as by promoting the proliferation of smooth muscle, thickening vascular walls, and damaging renal podocytes [10]. Therefore, considering the pathological overlap in sphingolipidoses and other LSDs, we decided to measure Lyso-Gb3 cross-sectionally in LSD model cells with uniform genetic backgrounds and in patient specimens to clarify the profile of Lyso-Gb3 accumulation.
2. Results and Discussion
To systematically compare whether lysosomal dysfunction in various LSDs affects lipid metabolism and leads to Lyso-Gb3 accumulation, we established disease model cells by knockout (KO) of 25 lysosomal enzymes or lysosome-related genes using CRISPR/Cas9 in the HeLa cell line: ARSA KO (metachromatic leukodystrophy), ARSB KO (mucopolysaccharidosis VI), ASAH1 KO (Farber disease), CTSA KO (galactosialidosis), CTSD KO (neuronal ceroid lipofuscinosis 10), FUCA1 KO (fucosidosis), GAA KO (Pompe disease), GALC KO (Krabbe disease), GBA KO (Gaucher disease), GLA KO (Fabry disease), GLB1 KO (GM1 gangliosidosis), GNPTAB KO (ML II/III), GUSB KO (mucopolysaccharidosis VII), HEXA KO (Tay-Sachs disease), HEXB KO (Sandhoff disease), IDS KO (mucopolysaccharidosis II), LIPA KO (LAL-D), MAN2B1 KO (α-mannosidosis), MCOLN1 KO (mucolipidosis IV), NEU1 KO (sialidosis), NPC1 KO (Niemann-Pick disease type C), PPT1 KO (neuronal ceroid lipofuscinosis 1), PSAP KO (combined saposin deficiency), SMPD1 KO (Niemann-Pick disease type A/B), and TPP1 KO (neuronal ceroid lipofuscinosis 2). We collected cell pellets and quantified Lyso-Gb3 in the cells by LC-MS/MS. The results showed that Lyso-Gb3 levels were more than three times higher than those in non-edited cells in three cell lines, including GLA KO, GNPTAB KO, and PSAP KO cells. These cells showed a statistically significant or a tendency to increase in Lyso-Gb3 compared to non-edited cells. NEU1 KO and NPC1 KO cells showed approximately two-fold increases, and the other cell lines showed no obvious increase in Lyso-Gb3 compared to non-edited cells (Figure 1A).
Lyso-Gb3 has already been established as a useful clinical biomarker for Fabry disease [7,8,9]. Therefore, the detection of increased Lyso-Gb3 in the GLA KO cells demonstrates the validity of the model cell and the measuring system.
Prosaposin, encoded by the PSAP gene, is the precursor of saposins A, B, C, and D, which are produced by post-translational cleavage [11,12]. Deficiency of PSAP causes combined saposin deficiency, which is a fatal infantile storage disorder with hepatosplenomegaly and severe neurologic symptoms [12,13]. Each saposin is a co-factor for the hydrolytic reaction of lysosomal enzymes. Because SapB supports the degradative functions of α-galactosidase A (encoded by GLA), it is conceivable that the loss of prosaposin caused the intracellular accumulation of Lyso-Gb3 [14].
ML II/III is caused by a biallelic mutation in GNPTAB [15,16,17], leading to a defect of GlcNAc 1-phosphotransferase that works for the addition of the M6P residues on lysosomal enzymes. M6P residues are necessary for the targeting of lysosomal enzymes to lysosomes [18]. Since α-galactosidase A (encoded by GLA) and lysosomal acid lipase (encoded by LIPA) are also transported to lysosomes via the M6P-dependent pathway, M6P-modified enzymes are used in enzyme replacement therapy for Fabry and LAL-D. It is speculated that Lyso-Gb3 accumulation in ML II cells is caused by a combined effect of a lack of lysosomal enzymes and the disruption of acidic environments inside lysosomes [2].
The amounts of Lyso-Gb3 were investigated in ML II/III patient specimens. Lyso-Gb3 was significantly elevated in ML II/III patient skin fibroblasts compared with normal skin fibroblasts (Figure 1B). The cellular accumulation of Lyso-Gb3 showed a tendency to decrease (p = 0.097) after total lysosomal enzyme supplementation (Figure 1B). These results indicate that the levels of Lyso-Gb3 have biomarker properties that change in response to treatment.
Lyso-Gb3 in patient plasma was measured from residual specimens used for the diagnosis of LSDs. Among the diseases targeted in Figure 1A, LSDs, for which multiple measurement data were obtained, are shown in Figure 1C. Although an increase in Lyso-Gb3 was observed in some Fabry disease patients’ plasma, no clear increase in Lyso-Gb3 was observed in other LSDs measured in this study, such as Gaucher, Krabbe, metachromatic leukodystrophy (MLD), and ML II/III. These data are consistent with a previous report [19]. Due to the limited number of samples analyzed, further analysis is awaited to reach a conclusion. Interestingly, Lyso-Gb3 does not appear to leak into the plasma of ML II/III patients (Figure 1C), although cellular accumulation of Lyso-Gb3 is comparable between the Fabry and ML II/III models. The difference in plasma Lyso-Gb3 in patients with Fabry disease and ML II/III has important implications for the pathomechanism of these LSDs.
Recently, measurements of lysosomal enzyme activity in lymphocytes in dried blood spots have been used in newborn screening of LSDs. B lymphocytes in ML II have been found to have an accumulation of storage material and impaired function [20]. Our results suggest that measurements of Lyso-Gb3 in blood cells may be a biomarker for ML II/III, even though Lyso-Gb3 does not change in plasma.
3. Materials and Methods
3.1. Knockout (KO) Cell Lines
Lysosome-related genes were knocked out using CRISPR/Cas9 in HeLa Kyoto, a cell line for which whole genome analysis has been performed [21]. HeLa Kyoto cells were originally established at Kyoto University based on canonical HeLa cells, were provided to EMBL, and are now distributed worldwide without any limitation (RRID: CVCL_1922). Plasmids expressing each CRISPR guide RNA and Cas9 protein were prepared based on px458 (pSpCas9(BB)-2A-GFP (PX458)), which was a gift from Feng Zhang (Addgene plasmid #48138; http://n2t.net/addgene:48138 (accessed on 3 October 2017); RRID: Addgene_48138) [22]. Guide sequences for gene targeting were designed using online tools, either the Benchling CRISPR Guide RNA Design Tool (https://www.benchling.com/crispr/ (accessed on 6 July 2020)) or CRISPRdirect (https://crispr.dbcls.jp/ (accessed on 24 June 2022)) (Table 1). The double-stranded DNA coding guide RNA sequences were prepared by annealing synthesized complementary DNA oligo pairs on a thermal cycler after phosphorylation by the T4 polynucleotide kinase (TaKaRa Bio, Shiga, Japan) and then cloned at the BbsI site of the px458 plasmid. The targeting plasmid was transfected into HeLa Kyoto cells with the Effectene Transfection reagent (Qiagen, Hilden, Germany). After 48 h of transfection, GFP-positive cells were sorted into 96-well plates as a single cell culture by FACSAria III™ Cell Sorter (BD Biosciences, CA, USA) and expanded. Genomic DNA from each clone was prepared using the QuickExtract™ DNA Extraction Solution (Lucigen, Oxford, UK). The genomic sequence of each targeted site was PCR-amplified with gene-specific primer sets, and genome editing was analyzed by Sanger sequencing. KO clones were characterized by phenotyping either lysosomal enzyme activities, substrate accumulation, or autophagic flux and confirmed by genotyping. The established LSD model cell lines were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Merck, MA, USA) supplemented with 10% fetal bovine serum FBS, 100 units/mL penicillin, and 100 µg/mL streptomycin (Nacalai-Tesque, Kyoto, Japan). The cell pellets for LC-MS/MS analysis were prepared by harvesting cells from multiple dishes in subconfluent conditions with trypsin–EDTA digestion, followed by several PBS(-) washes.
3.2. Patient Specimens
ML II/III patient skin fibroblasts (SFs) and LSD patient plasma were obtained from residual specimens of diagnostic research after obtaining informed consent. The use of the patients’ specimens was approved by the institutional ethics review board (Kawasaki Medical School: approval Number: 5889). Patient SF lines were established from a 5 mm square of buttock skin piece from each patient. The tissue was excised by punching and then shredded with a clean scalpel and scissors. The disrupted skin tissue was directly transferred and cultured in the AmnioMAX-II™ medium (Thermo Fisher, Waltham, MA, USA). After weeks of expansion, grown colonies were dispersed by trypsin–EDTA digestion and thereafter cultured in the normal DMEM as described above in the HeLa Kyoto part. Genotypes of ML II/III patient SFs were #4 c.310C>T (p.Q104X)/c.2522delA (p.K848fs), #5, and #6 c.1120T>C (p.F374L)/c.3565C>T (p.R1189X). For healthy controls, three commercially available normal SFs were purchased from Kurabo (Kurashiki, Japan), Thermo Fisher Scientific Inc. (Waltham, MA, USA), and Lonza (Basel, Switzerland).
In vitro therapeutic intervention for ML II/III SFs via total lysosomal enzyme supplementation was performed as described before [2]. In short, the normal SF was incubated with ammonium chloride, and M6P-tagged lysosomal enzymes exhaled in the culture supernatant were collected. Ammonium chloride was removed using a molecular weight cut-off filter, and the lysosomal enzyme mixture was purified. ML II/III patient SFs were treated with a conditioned medium containing the M6P-tagged lysosomal enzyme mixture for 72 h to replenish lysosomal enzymes.
3.3. LC-MS/MS Analysis for Lyso-Gb3
The internal standard reagent was purchased from Avanti Polar Lipid (Alabaster, CA, USA). Methanol, isopropanol, and chloroform of ultra-performance liquid chromatography (UPLC)/MS quality were obtained from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Ultrapure water was obtained from a Milli-Q water system (Millipore, Billerica, MA, USA). Briefly, cell lines were mixed with 1.0 mL of methanol containing the internal standards. The samples were briefly sonicated and incubated on ice for 0.5 h. The samples were then centrifuged for 3 min at 10,000× g. Liquid phases were collected in measuring vials. LC-MS/MS analysis was performed using the Xevo TQ-XS mass spectrometer with an ACQUITY UPLC H-Class system (Waters). The lipids were separated on a Waters X-Bridge C18 column (3.5 mm, 150 mm × 1.0 mm internal diameter) at 40 °C using a gradient solvent system as follows: mobile phase A was isopropanol/methanol/water (5/1/4 v/v/v) supplemented with 5 mM ammonium formate and 0.05% ammonium hydroxide (28% in water); mobile phase B was isopropanol supplemented with 5 mM ammonium formate and 0.05% ammonium hydroxide (28% in water) with a flow rate of 80 mL/min. Lyso-Gb3 metabolites were measured using multiple reaction monitoring (MRM) in the positive ion mode. The peak areas of the individual species were normalized to those of internal/surrogate standards, which were added to the samples before lipid extraction. Raw LC-MS/MS data were processed using analytical software (MassLynx 4.2; Waters). The quantification and annotation methods used in this study correspond to “absolute quantification Level 2” and the “Fatty Acyl/Alkyl Level“ defined by the Lipidomics Standard initiative, respectively [23].
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Platt F.M. d’Azzo A. Davidson B.L. Neufeld E.F. Tifft C.J. Lysosomal storage diseases Nat. Rev. Dis. Primers 201842710.1038/s 41572-018-0025-430275469 · doi ↗ · pubmed ↗
- 2Otomo T. Higaki K. Nanba E. Ozono K. Sakai N. Lysosomal storage causes cellular dysfunction in mucolipidosis II skin fibroblasts J. Biol. Chem.2011286352833529010.1074/jbc.M 111.26793021846724 PMC 3186395 · doi ↗ · pubmed ↗
- 3Moriwaki T. Terawaki S. Otomo T. Impaired lysosomal acidity maintenance in acid lipase-deficient cells leads to defective autophagy J. Biol. Chem.202430010574310.1016/j.jbc.2024.10574338354786 PMC 10933554 · doi ↗ · pubmed ↗
- 4Kondo H. Maksimova N. Otomo T. Kato H. Imai A. Asano Y. Kobayashi K. Nojima S. Nakaya A. Hamada Y. Mutation in VPS 33A affects metabolism of glycosaminoglycans: A new type of mucopolysaccharidosis with severe systemic symptoms Hum. Mol. Genet.20172717318310.1093/hmg/ddw 37728013294 · doi ↗ · pubmed ↗
- 5Cyske Z. Gaffke L. Pierzynowska K. Węgrzyn G. Mucopolysaccharidosis-Plus Syndrome: Is This a Type of Mucopolysaccharidosis or a Separate Kind of Metabolic Disease?Int. J. Mol. Sci.202425957010.3390/ijms 2517957039273517 PMC 11395409 · doi ↗ · pubmed ↗
- 6Desnick R.J. Ioannou Y.A. Eng C.M. The Online Metabolic and Molecular Bases of Inherited Disease, Part 16: Lysosomal Disorders, α-Galactosidase A Deficiency: Fabry Disease Available online: https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225546984(accessed on 20 June 2025)
- 7Aerts J.M. Groener J.E. Kuiper S. Donker-Koopman W.E. Strijland A. Ottenhoff R. van Roomen C. Mirzaian M. Wijburg F.A. Linthorst G.E. Elevated globotriaosylsphingosine is a hallmark of Fabry disease Proc. Natl. Acad. Sci. USA 20081052812281710.1073/pnas.071230910518287059 PMC 2268542 · doi ↗ · pubmed ↗
- 8Maruyama H. Miyata K. Mikame M. Taguchi A. Guili C. Shimura M. Murayama K. Inoue T. Yamamoto S. Sugimura K. Effectiveness of plasma lyso-Gb 3 as a biomarker for selecting high-risk patients with Fabry disease from multispecialty clinics for genetic analysis Genet. Med.201821445210.1038/gim.2018.3129543226 PMC 6363642 · doi ↗ · pubmed ↗