NsrR Represses σE-Dependent Small RNAs and Interacts with RpoE via a Noncanonical Mechanism in Escherichia coli
Joseph I. Aubee, Jalisa Nurse, Dale Lewis, Chin-Hsien Tai, Karl M. Thompson

TL;DR
This study reveals that NsrR, a nitric oxide sensor, represses σE activity in E. coli, influencing envelope stress responses through a novel mechanism.
Contribution
NsrR is shown to act as a noncanonical anti-sigma-like regulator of σE, independent of RseA.
Findings
NsrR represses σE-dependent small RNAs like rybB, micA, and micL.
NsrR interacts directly with σE, as shown by bacterial two-hybrid assays.
NsrR's repression of σE activity occurs with kinetics similar to RseA.
Abstract
The envelope stress response in Escherichia coli is primarily governed by the sigma factor RpoE (σE), which activates protective genes upon membrane perturbation. Under non-stress conditions, σE is sequestered by its anti-sigma factor RseA. In this study, we identify an unexpected role for the nitric-oxide-sensing repressor NsrR in dampening σE activity and repressing σE-dependent small RNAs, including rybB, micA, and micL. Overexpression of nsrR represses transcription from σE-dependent promoters and phenocopies σE inactivation, resulting in filamentous morphology and growth defects. Conversely, ΔnsrR de-represses σE targets, with additive effects in rseA mutants—supporting an RseA-independent regulatory role. Time-course analysis shows NsrR represses σE activity, with kinetics comparable to those of RseA. While in vitro assays failed to detect robust NsrR binding to σE target…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
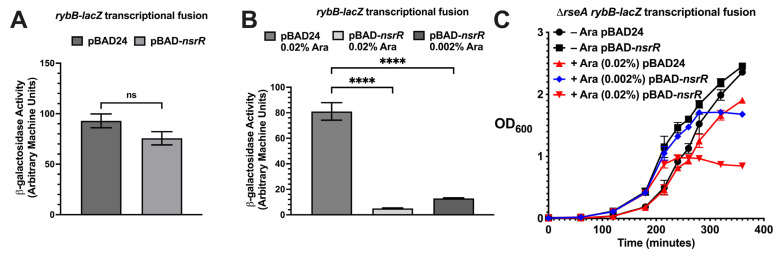 Figure 1
Figure 1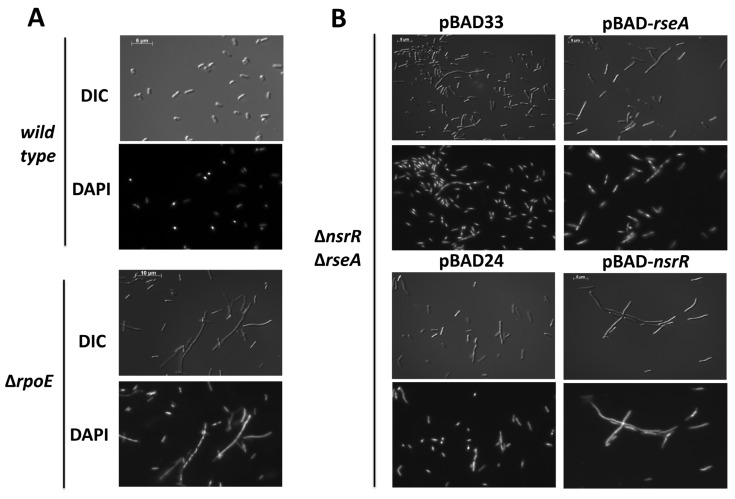 Figure 2
Figure 2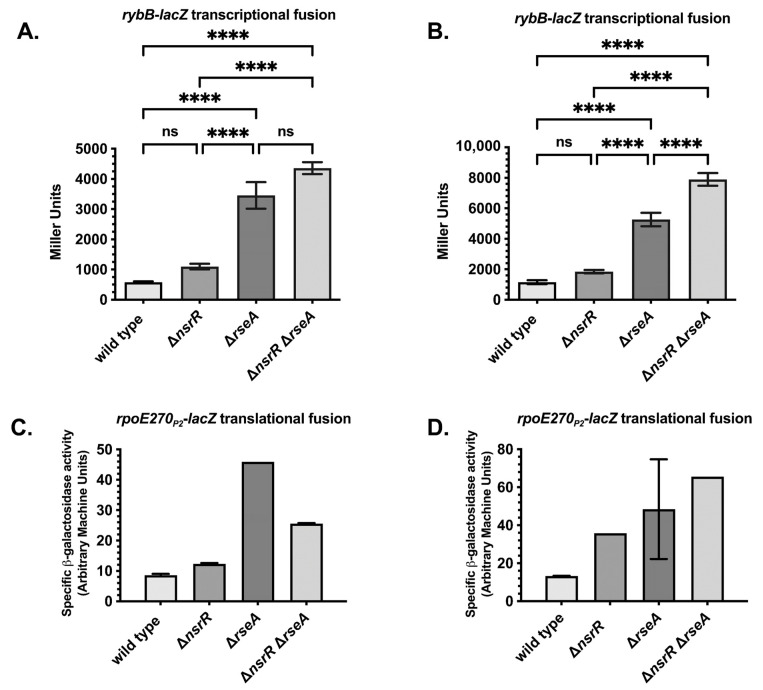 Figure 3
Figure 3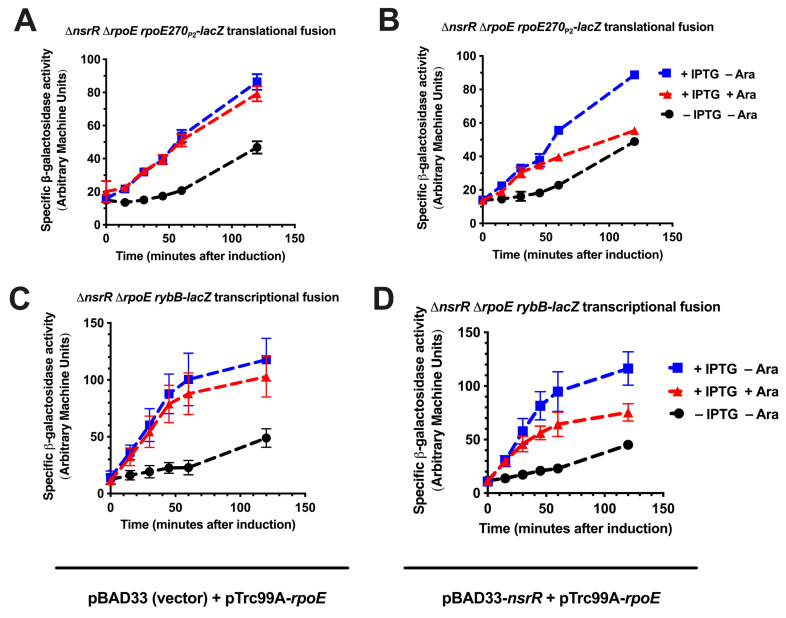 Figure 4
Figure 4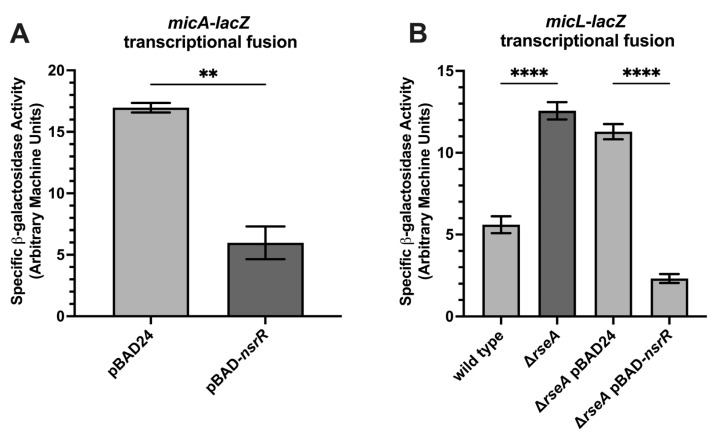 Figure 5
Figure 5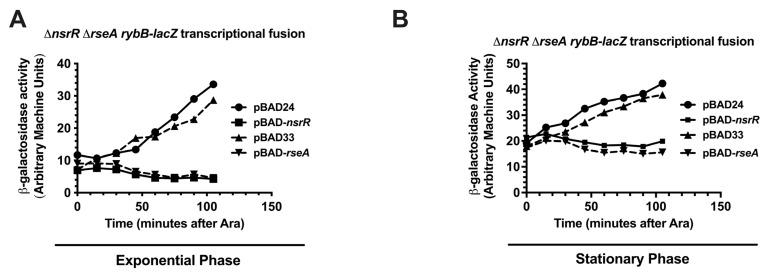 Figure 6
Figure 6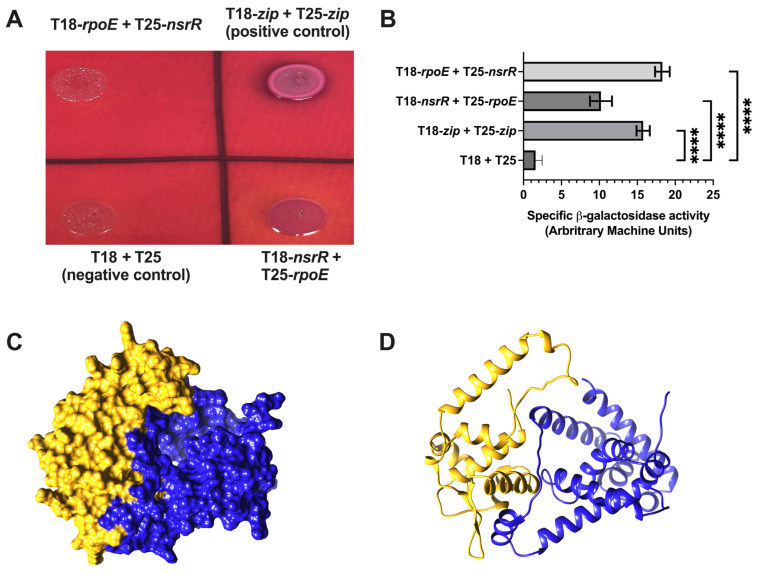 Figure 7
Figure 7- —National Institute of General Medical Sciences
- —National Science Foundation (NSF)
- —graduate student support
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial Genetics and Biotechnology · RNA and protein synthesis mechanisms · Gene Regulatory Network Analysis
1. Introduction
RpoE (σ^E^) is an extracytoplasmic function (ECF) sigma factor that governs the envelope stress response in Escherichia coli and many other Gram-negative bacteria [1,2,3]. Under non-stress conditions, RpoE is sequestered at the inner membrane by direct binding to its anti-sigma factor (RseA) [4,5]. Activation of the σ^E^ regulon requires the proteolytic degradation of RseA, a process triggered by envelope stress signals, such as misfolded or accumulating outer membrane proteins (OMPs) [2,6,7,8]. These signals initiate a regulated intramembrane proteolysis (RIP) cascade, in which DegS and RseP sequentially cleave RseA, the anti-sigma factor that sequesters σ^E^ to the inner membrane, followed by ClpXP-mediated degradation of the remaining cytoplasmic fragment [4,6,9,10,11]. This cascade releases RpoE to initiate the transcription of target genes involved in maintaining envelope integrity, including chaperones, proteases, and small regulatory RNAs (sRNAs) [12,13,14,15,16,17,18,19,20,21,22].
Among the most critical outputs of the σ^E^ response are sRNAs that downregulate OMP synthesis, thereby reducing the burden on the envelope and helping to restore homeostasis [13,15,23,24]. Key members of this group include MicA, MicL, and RybB, all of which repress the translation of OMP-encoding mRNAs [15,18,25,26,27]. These sRNAs function in negative feedback loops that limit RpoE activation and fine-tune the envelope stress response. RybB, an 80-nucleotide sRNA under the direct control of RpoE, has been shown to target multiple OMPs and participate in ppGpp-mediated stress adaptation [13,15,20,24,28,29,30,31,32].
During a genetic screen for novel repressors of rybB, we identified a multicopy clone containing nsrR, a nitric oxide (NO)-responsive transcriptional repressor [15,33,34]. Overexpression of nsrR was sufficient to suppress both the expression of rpoE and rybB, even in the absence of RseA, suggesting an RseA-independent mechanism of repression [15]. These findings raised the possibility that NsrR may influence the σ^E^ response through an uncharacterized regulatory interaction. NsrR is a conserved NO-sensing transcriptional repressor that plays central roles in oxidative stress defense and NO detoxification in diverse bacterial species [33,34]. Chromatin immunoprecipitation and microarray analyses (ChIP-chip) have identified dozens of direct NsrR targets in E. coli, including genes involved in motility and biofilm regulation [35]. However, rpoE and rybB were not among the identified NsrR-bound promoters, suggesting that their repression may occur via an indirect mechanism [35].
To clarify how NsrR represses rpoE and rybB, we considered three potential mechanisms: (1) indirect repression of rybB via reduced rpoE expression, (2) inhibition of RpoE activity through anti-sigma-like interactions, and (3) direct repression of both genes by a mechanism distinct from NsrR’s NO-regulated regulon. Herein, we show that NsrR represses RpoE-dependent transcription, with kinetics and strength comparable to those of RseA. NsrR overexpression blocks RpoE autoregulation and downstream target induction, even when RpoE is expressed from a plasmid. Moreover, NsrR overexpression phenocopies ΔrpoE, producing filamentous cell morphology and impaired growth. We further demonstrate that NsrR physically interacts with RpoE in bacterial two-hybrid assays, and AlphaFold3 modeling predicts a plausible interaction interface [36]. To dissect these mechanistic models, we employed a combination of genetics, transcriptional reporters, two-hybrid interaction assays, and structural prediction to test whether NsrR modulates σ^E^ activity through direct interaction, indirect repression, or both. Together, these findings support a model in which NsrR acts as a noncanonical modulator of RpoE, functioning outside its traditional NO-responsive role to shape the envelope stress response.
2. Results
2.1. NsrR Overexpression Represses σE-Dependent Promoters and Disrupts Cell Division
As previously reported, overproduction of NsrR was sufficient to repress rybB-lacZ and rpoE270P2-lacZ fusion in the absence of rseA [15]. NsrR was expressed from an arabinose-inducible promoter, and the addition of arabinose was necessary to see repression (Figure 1A,B; 1A without arabinose and 1B with arabinose). The overexpression of nsrR in the presence of 0.02% arabinose resulted in a severe growth defect (Figure 1C); the overexpression of nsrR in the presence of 0.002% arabinose did not inhibit growth as rapidly (Figure 1C). These growth defects prompted us to hypothesize that NsrR impairs cell division or chromosome segregation. A previous report demonstrated aberrant cellular morphology phenotypes upon inhibition of σ^E^ activity because of RseA overexpression [37]. We hypothesized that we would observe aberrant cellular morphology because of NsrR overexpression, due to the NsrR repression of rpoE. Therefore, we examined the cellular morphology upon the overexpression of NsrR, using RseA overexpression and the absence of rpoE as positive controls. For our positive control, wild-type and ∆rpoE cells were grown to the mid-exponential phase (OD_600_ = 0.5), and aliquots of the cultures were then harvested and analyzed by differential interference contrast and fluorescence microscopy using DAPI staining. The absence of rpoE led to severe cellular filamentation in a significant proportion of the population of cells (Figure 2A). For the analysis of the NsrR overexpression, we transformed a ∆nsrR ∆rseA double mutant with a plasmid containing an arabinose-inducible allele of rseA or nsrR. We then grew the cells in LB supplemented with ampicillin or chloramphenicol and 0.002% arabinose to an OD_600_ of 0.7–1.0. We then harvested aliquots of these cultures and analyzed them by differential interference contrast and fluorescence microscopy using DAPI staining (Figure 2B). Consistent with previous reports [37], overexpression of RseA leads to the formation of membrane blebs (Figure 2B). In addition, there was also some cellular filamentation in a proportion of the cells. The overexpression of nsrR led to a severe cellular filamentation that was seen in many of the cells (Figure 2B). Taken together, this suggests that nsrR overexpression results in cellular morphology defects that are very similar to those seen upon removing or inactivating rpoE.
2.2. NsrR and RseA Independently Repress rybB and rpoE, Supporting a Parallel Regulatory Model
To assess the physiological significance of the effects of nsrR on rybB and rpoE, we tested the effect of ΔnsrR on the expression of rybB and rpoE. We transduced ΔnsrR::tet mutations into strains carrying rybB-lacZ transcriptional fusions and rpoE270P2-lacZ translational fusions, in wild-type or ΔrseA genetic backgrounds, and measured their activity at various stages of growth. As expected, ΔrseA significantly increased the expression levels of both fusions, both in the exponential (Figure 3A,C) and stationary phases of growth (Figure 3B,D). The absence of nsrR resulted in an approximate 2-fold increase in rybB-lacZ expression in rseA^+^ cells, which was statistically significant, in both the exponential and stationary phases of growth (Figure 3A,B). The statistically significant increase in the expression of rybB-lacZ in rseA mutants is much greater at approximately 7-fold in the exponential phase and 4-fold in the stationary phase (Figure 3A,B). In cells deleted for rseA, ΔnsrR further increased the activity of rybB-lacZ (Figure 3A,B), confirming and extending our previous finding that NsrR repression is independent of RseA. However, this increase was not statistically significant in exponential-phase cultures. The ΔnsrR mutant strain of rpoE270P2-lacZ also had a statistically significant increase in expression versus the wild-type strain (Figure 3C,D). While this increase was less than 2-fold in the exponential phase, there was a 2–3-fold increase in the stationary phase (Figure 3C,D). In contrast with the rybB-lacZ fusion strain, the ΔnsrR ΔrseA double mutant did not exhibit an increase in activity in comparison to the ΔrseA mutant in exponential phase for reasons that are not clear. In stationary phase, there is a slight additive increase that is not likely to be statistically significant (Figure 3C,D). Overall, these results confirm that the multicopy repression previously seen reflects a physiologically relevant role of NsrR in the regulation of the σ^E^ response.
2.3. NsrR Interferes with RpoE-Dependent Promoter Activation in an Inducible Expression System
To test this hypothesis, we measured the ability of NsrR to repress rpoE or rybB promoters in strains in which the sole source of rpoE and nsrR was from compatible plasmids with different inducible promoters (on the pTrc99A-rpoE and pBAD33-nsrR plasmids, respectively). To execute the experiment, we grew the cultures to an OD_600_ of 0.5 and then added IPTG or IPTG and arabinose and isolated samples every 15 min after induction for 120 min (Figure 4). For both the rpoE270P2-lacZ translational and rybB-lacZ transcriptional fusions, the fusions were active in the absence of IPTG and increased over time, suggesting that there is leaky induction of rpoE from the plasmid-borne IPTG-inducible allele of rpoE (Figure 4A–D, black line). Upon the addition of IPTG, the activities of both the rpoE270P2-lacZ translational and rybB-lacZ transcriptional fusions increased over time and were significantly greater than the activities in the absence of IPTG, confirming the σ^E^-dependent activation of rpoE270P2-lacZ and rybB-lacZ, as expected (Figure 4A–D, blue line). This induction was unaffected by the induction of the pBAD33 vector with arabinose (Figure 4A,C, red line). However, the induction of NsrR decreased the activities of both the rpoE270P2-lacZ and rybB-lacZ fusions, in comparison to the activities of the fusions in the presence of IPTG alone (compare red to blue lines in Figure 4B,D). Therefore, overexpression of NsrR can inhibit the σ^E^ induction of rybB and rpoEP2 promoters and is not simply acting at the level of RpoE synthesis. This would presumably occur through the direct interaction of NsrR with either the σ^E^-dependent rybB and rpoEP2 promoters through interaction with the σ^E^ protein itself in a manner analogous to RseA.
2.4. NsrR Represses Additional σE-Regulated sRNAs
To determine whether NsrR represses additional σ^E^-regulated small RNAs beyond rybB, we examined micA and micL expression using lacZ transcriptional reporter fusions. The micA-lacZ strain harboring either pBAD24 or pBAD24-nsrR was grown to the late exponential phase of growth in LB supplemented with 0.02% arabinose. Overexpression of nsrR significantly reduced the micA-lacZ activity (Figure 5A). Similarly, nsrR overexpression repressed the micL-lacZ expression in a ΔrseA background (Figure 5B), suggesting that this repression occurs independently of RseA-mediated sequestration. These findings extend NsrR’s regulatory reach within the σ^E^ regulon and raise the possibility that, like RseA, NsrR may function in a parallel pathway to modulate σ^E^ activity. This observation prompted us to compare repression kinetics of NsrR and RseA on the rybB promoter.
2.5. NsrR Does Not Exhibit Robust Binding to σE Target Promoters In Vitro
To test whether NsrR directly represses rpoE or rybB through promoter binding, we performed electrophoretic mobility shift assays (EMSAs), DNase I footprinting, and in vitro transcription assays using purified NsrR and radiolabeled promoter fragments. However, none of these approaches revealed consistent or robust DNA-binding activity under standard conditions. These negative results suggest that NsrR may not repress these promoters via classical DNA binding or that such interactions are weak, transient, or context dependent—potentially requiring stress signals or co-factors absent in vitro. These findings are consistent with previous ChIP-seq studies that did not identify rpoE or rybB as direct NsrR targets [38]. To address this possibility, we plan to apply recently developed chromatin-profiling methods with significantly enhanced sensitivity over the previous technical standard (ChIP-seq), under defined stress conditions, as described in the discussion, to evaluate the potential for condition-dependent promoter occupancy.
2.6. NsrR Phenocopies RseA Repression Kinetics at the rybB Promoter
To test the relative repression kinetics of RseA and NsrR on rybB transcription (as a representative of the σ^E^ regulon with one of the strongest promoters), we activated the rybB-lacZ transcriptional fusion followed by the rapid induction of NsrR and RseA—measuring β-galactosidase activity every 15 min for 105 min. The rationale behind this experiment is that if NsrR directly represses σ^E^-dependent promoters, the kinetics of NsrR repression of rybB will be similar or identical to RseA repression. If NsrR acts indirectly (represses or activates the synthesis of another regulator, for instance), we would expect NsrR repression to be significantly slower than repression by the RseA anti-sigma factor, reflecting the time to either deplete or accumulate the direct regulator. To tightly control nsrR and rseA levels for this assay, we used a rybB-lacZ transcriptional fusion strain with a ΔnsrR ΔrseA genetic background transformed with vectors or with plasmids carrying arabinose-inducible alleles of nsrR or rseA. We then grew these strains and the vector controls in LB supplemented with 0.2% glucose to an OD_600_ of 0.5 (exponential—Figure 6A) or 1.0 (stationary—Figure 5B) and shifted the cells to LB supplemented with 0.2% arabinose. We analyzed culture aliquots for specific β-galactosidase activity at 15 min intervals, following arabinose induction, up to 120 min. For the cultures shifted in exponential phase, the specific β-galactosidase activity of the rybB-lacZ transcriptional fusion strain containing the pBAD24 or pBAD33 vector controls increased over time, consistent with the activity of σ^E^-dependent promoters. The activity in the strain containing pBAD33-rseA did not increase, and slightly decreased, consistent with the known direct negative regulation of σ^E^ activity by RseA. The strain containing pBAD24-nsrR was very similar in behavior to the RseA-expressing strain. These kinetics suggest that NsrR directly regulates σ^E^-dependent transcription. Taken together, these observations prompted us to investigate a potential NsrR-RpoE protein–protein interaction mechanism.
2.7. NsrR–RpoE Interaction Confirmed by Bacterial Adenylate Cyclase Two-Hybrid (BACTH) and Explored via AlphaFold3 Multimer Modeling
Given the rapid and parallel repression kinetics of NsrR and RseA at the rybB promoter, we hypothesized that NsrR may regulate σ^E^ activity post-translationally through a direct interaction with RpoE, analogous to the canonical anti-sigma factor (RseA). We employed the BACTH system to test for interactions and used AlphaFold3 multimer modeling to visualize potential interaction surfaces. The BACTH system detects protein–protein interactions based on cAMP-dependent transcriptional activation. Reciprocal fusions of NsrR and RpoE to the T18 and T25 adenylate cyclase fragments produced detectable colony color on MacConkey maltose agar comparable to that of the leucine zipper positive control (Figure 7A). Quantitative β-galactosidase assays confirmed a significant interaction between NsrR and RpoE relative to vector-only controls (Figure 7B). To investigate the molecular nature of this interaction, we used AlphaFold3 multimer modeling, which offers improved capabilities for predicting multi-subunit complexes and flexible protein interfaces, to model the NsrR–RpoE interaction [36]. Direct modeling of the NsrR–RpoE heterodimer yielded low confidence scores (pTM 0.48, ipTM 0.17; Figure S1A), suggesting model uncertainty. However, consistent with prior reports that NsrR functions as a dimer [39], AlphaFold3 confidently predicted the NsrR homodimer (pTM 0.83, ipTM 0.82; Figure 7C). We then modeled the interaction between the NsrR dimer and RpoE. Among five predicted models (Figure 7D and Supplemental Figure S1B–E), the NsrR dimer structure was stable, but the RpoE orientation varied, likely due to the presence of a flexible linker region (residues 92–116). When RpoE is a part of the RNA polymerase holoenzyme (e.g., in complex with RpoA, RpoB, RpoC, and RpoZ; PDB: 6JBQ) [40], this linker is extended, allowing its σ^70^ domains to contact promoter DNA. In contrast, when RpoE is bound to RseA (PDB: 1OR7—Supplemental Figure S1G) [40], the linker adopts a helical structure, bringing its N- and C-terminal domains into close proximity. These differences highlight the challenge of predicting flexible domain orientations with current algorithms. While AlphaFold3 offers valuable structural insight, experimental validation through mutagenesis or high-resolution structural methods will be necessary to confirm the specific mode of the NsrR–RpoE interaction.
3. Discussion
3.1. NsrR Is a Dual Regulator of Envelope and Nitrosative Stress Responses
Our study reveals an expanded role for the nitric oxide (NO)-sensing transcriptional regulator (NsrR) in Escherichia coli, extending beyond its canonical repression of NO detoxification and iron–sulfur cluster maintenance genes. We demonstrate that NsrR modulates the σ^E^ (RpoE) envelope stress response by repressing σ^E^-regulated targets, including rybB, micA, micL, and rpoE itself. These effects occur independently of RseA, the membrane-bound anti-sigma factor that typically sequesters σ^E^ under non-stress conditions, suggesting that NsrR functions through a parallel regulatory mechanism.
Rather than constraining σ^E^ under basal conditions—when it is already sequestered—NsrR may act after the initial activation of σ^E^, functioning as a secondary brake that dampens prolonged or excessive envelope stress signaling. This dual role positions NsrR as a redox-sensitive checkpoint that coordinates nitrosative and envelope stress responses. By modulating σ^E^ output following activation, NsrR helps to maintain the balance between outer membrane protein (OMP) synthesis and envelope remodeling during cell elongation and division—an essential equilibrium for bacterial viability.
3.2. RybB Repression as a Model for the NsrR-Dependent Modulation of σE
We used the small RNA rybB, one of the most prominent σ^E^ targets, as a model for dissecting the NsrR-dependent modulation of the envelope stress response. NsrR overexpression strongly repressed rybB-lacZ activity and phenocopied σ^E^ inactivation, producing filamentous morphology and growth defects. Conversely, ΔnsrR resulted in elevated expression of both rybB and rpoE, with additive effects in a ΔrseA background. This suggests that NsrR and RseA exert independent yet convergent repression of σ^E^ activity.
Importantly, NsrR also repressed micA and micL, two additional σ^E^-regulated sRNAs, highlighting a broader role in the post-transcriptional branch of the σ^E^ regulon. These sRNAs fine-tune OMP levels by promoting the degradation of outer membrane protein mRNAs. Thus, NsrR’s repression of these regulatory RNAs suggests it serves not only as a transcriptional repressor but also as a master regulator that constrains downstream RNA-based stress adaptations to limit energy expenditure and prevent deleterious remodeling of the cell envelope.
3.3. NsrR May Act as a Noncanonical Anti-σE Factor
Given the kinetic and phenotypic similarities between NsrR and RseA repression of σ^E^-dependent promoter activity, we hypothesized that NsrR could directly modulate σ^E^ activity via protein–protein interactions. Supporting this model, bacterial two-hybrid assays revealed a direct interaction between NsrR and RpoE, further corroborated by multimer modeling (Figure 6), which predicted a plausible NsrR–RpoE interface. These data support a model in which NsrR may act as a noncanonical anti-σ^E^ factor, directly antagonizing the σ^E^ function through physical sequestration or inhibition.
Such dual-function regulators—combining transcriptional repression with direct sigma factor binding—are rare but increasingly recognized in bacterial stress networks. This model draws conceptual parallels to the Bacillus subtilis SigB system, where a complex cascade of sigma, anti-sigma, and anti-anti-sigma factors modulates stress responses [41,42]. In E. coli, a redox-sensitive interaction between NsrR and σ^E^ could serve as a novel regulatory checkpoint, integrating environmental NO signals with envelope stress signaling.
3.4. Physiological and Evolutionary Implications
The ability of NsrR to repress σ^E^ targets may reflect an adaptive mechanism that prioritizes NO detoxification and redox homeostasis over envelope repair during transient stress. In host environments, such as macrophages—where NO exposure and membrane perturbation often co-occur—this regulatory logic ensures that envelope stress responses are only activated when both redox and envelope stress thresholds are surpassed. Under non-lethal conditions, NsrR-mediated repression may help to avoid the futile or energetically costly induction of envelope repair pathways.
Upon the oxidation of NsrR’s [Fe–S] cluster, de-repression of the σ^E^ regulon would allow the appropriate induction of protective and reparative functions. This model supports a broader framework in which NsrR functions not only as a sensor of NO but also as a temporal gatekeeper for multi-stress adaptation, fine-tuning bacterial survival strategies under compound environmental challenges.
3.5. Open Questions and Future Directions
These findings suggest that NsrR may regulate σ^E^ through both direct protein–protein interactions and indirect regulatory effects, extending its role beyond the nitric oxide response. While the precise mechanisms remain to be defined, our data support a model in which NsrR functions as a noncanonical anti-sigma factor. Future studies will be required to clarify the molecular interface, regulatory scope, and physiological triggers of this interaction.
Notably, it remains unclear whether envelope stress directly modulates NsrR expression or activity. Although NsrR is classically regulated by nitric oxide via the oxidation of its [Fe–S] cluster, envelope stress or altered σ^E^ activity may influence nsrR stability or activity. Supporting this, Nicoloff et al. (2017) [43] identified a point mutation in nsrR, in a screen for suppressors of high σ^E^ activity in a ΔrseA genetic background. While the functional consequence of this point mutation is unknown, it is possible that it could augment repression of σ^E^ synthesis or activity. Such a mechanism would position NsrR as both a redox-responsible regulator and a feedback inhibitor that retrains prolonged σ^E^ activity under envelope stress conditions.
Given the conservation of both NsrR and σ^E^ across Gram-negative bacteria, analogous cross-regulatory mechanisms may operate in pathogens such as Salmonella, Klebsiella, and Pseudomonas. Dissecting how NO-sensing regulators influence envelope integrity pathways could uncover novel strategies to disrupt bacterial stress tolerance, persistence, and virulence.
4. Materials and Methods
4.1. Media and Growth Conditions
E. coli strains were cultured in LB (Lennox, Richardson, TX, USA) medium or on LB agar plates with appropriate antibiotics and inducers as described. For plasmid selection and maintenance, antibiotics were added at the following final concentrations: kanamycin (25 μg/mL), chloramphenicol (25 μg/mL), tetracycline (25 μg/mL), and ampicillin (100 μg/mL). PBAD-regulated promoters were repressed by supplementing media with 0.2% glucose and induced with L-arabinose at final concentrations of 0.02% or 0.002%. IPTG was used at 10 or 100 μM for the induction of plasmid-encoded rpoE. Plasmids were introduced via TSS transformation or electroporation. P1vir phage transduction was used to transfer chromosomal mutations following standard protocols [44].
4.2. Strains, Plasmids, and Oligonucleotides
All the strains were derived from E. coli K-12 MG1655. Full lists of strains, plasmids, and primers are provided in Table 1, Table 2 and Table 3.
4.2.1. Chromosomal Deletion–Insertion Mutations of nsrR and rpoE
A ΔnsrR::tet mutation was generated by λ-Red recombineering [44,46,47,48]. A tetracycline resistance cassette flanked by 50 bp homology arms upstream and downstream of nsrR was amplified using primers KT237 and KT238 to create a linear allelic exchange substrate (AES) for recombination. The PCR product was electroporated into strain NM200 (mini-λ::cat) after the induction of λ-Red recombinase at 43.5 °C for 15 min [47]. Transformants (putative recombinants) were selected on LB–tetracycline plates and confirmed by PCR and sequencing. A ΔrpoE::kan mutation was generating in an identical manner with minor changes. Briefly, the AES was amplified using primers KT926 and KT927. And transformants (putative recombinants) were selected on LB-kanamycin plates.
4.2.2. Construction of Plasmid-Based Arabinose-Inducible nsrR and rseA Alleles
The nsrR gene was amplified from MG1655 genomic DNA, using primers KT191 and KT192, and digested with Eco RI and Pst I before ligation into pBAD24. Primers KT906 and KT907 were used for the PCR amplification of nsrR, which was digested with Sac I and Pst I before ligation into pBAD33. The rseA gene was amplified with primers KT95 and KT75 and digested with Sac I and Xba I and cloned similarly into pBAD33. The constructs were verified by restriction digestion and sequencing. The primers are listed in Table 3.
4.2.3. Construction of Bacterial Adenylate Cyclase Two-Hybrid Assay (BACTH) Plasmids
The nsrR and rpoE open reading frames were PCR-amplified using the gene-specific primers listed in Table 3 and cloned in-frame with either the T18 or T25 domain of adenylate cyclase. Cloning was performed using HiFi Assembly Master Mix (New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s instructions. The pEB355 and pEB354 vectors were used to generate T18 and T25 fusion constructs, respectively. Specifically, T18-nsrR was constructed using primers KT2223 and KT2224, and T18-rpoE was constructed using KT2225 and KT2226. T25-nsrR and T25-rpoE were constructed with primer pairs KT2227/KT2228 and KT2229/KT2230, respectively. All the plasmid constructs were confirmed by restriction digestion and Sanger sequencing and were subsequently used to assess NsrR–RpoE protein–protein interactions in the BACTH system.
4.3. Bacterial Two-Hybrid (BACTH) Assay
Protein–protein interactions were assessed using the bacterial adenylate cyclase two-hybrid (BACTH) system [49,50]. Plasmid constructs encoding in-frame fusions of nsrR or rpoE to the T18 or T25 domains of adenylate cyclase were introduced to E. coli BTH101 by electroporation. The following plasmid pairs were tested: pEB355 and pEB354 (empty vector (negative control)), pEB355-zip and pEB354-zip (positive control), pEB355-nsrR + pEB354-rpoE, and pEB355-rpoE + pEB354-nsrR. These constructs generated reciprocal T18 and T25 fusions of the two proteins of interest. Electrocompetent BTH101 cells were prepared by washing log-phase cultures three times with ice-cold sterile water. Plasmid mixtures were electroporated using the Bio-Rad MicroPulser™, Bio Rad, Hercules, CA, USA (Ec1 “Bacteria” setting), and cells were recovered in LB for 1 h at 30 °C. Aliquots were plated on LB agar containing ampicillin and kanamycin for the selection of double transformants. Selected colonies were grown overnight in liquid LB supplemented with the same antibiotics and 100 μM IPTG to induce the expression of the fusion proteins. Cultures were spotted onto MacConkey maltose agar plates and incubated at 30 °C for 24 to 48 h. The colony color was used as a qualitative measure of interaction-dependent cAMP restoration and lac operon activation. Quantitative β-galactosidase assays were performed, as described below, on parallel cultures to confirm and measure the interaction strength.
4.4. Quantitative β-Galactosidase Assays
β-galactosidase assays were performed using a standard protocol [51] or in 96-well plates, as previously described [52]. For the standard protocol, the specific β-galactosidase activity was expressed as Miller units, as previously described [51]. Cultures were harvested at the exponential or stationary phase, permeabilized, and incubated with ONPG. Absorbance at 420 nm measured using a SpectraMax 250 plate reader (Marshall Scientific, Hampton, NH, USA). The specific β-galactosidase activity was expressed as arbitrary machine units and defined as the slope of OD_420_/OD_600_. The results represent the means of at least three biological replicates.
4.5. Microscopy Experiments
Cells were grown to the mid-log phase of growth, fixed, and stained with DAPI. The morphology was analyzed using differential interference contrast (DIC) and fluorescence microscopy. Images were acquired on a Axio Imager.M2 upright fluorescence microscope (Carl Zeiss Microscopy, LLC, White Plains, NY, USA), using 100× magnification, and processed using Zeiss Zen software version 3.12 (Carl Zeiss Microscopy, LLC, White Plains, NY, USA).
4.6. Protein–DNA Interaction Assays
Electrophoretic Mobility Shift Assays (EMSAs), DNase I footprinting, and in vitro transcription assays were performed using purified NsrR, as well as RpoE reconstituted with purified RNA polymerase holoenzyme and radiolabeled promoter fragments from rpoE and rybB. Assays were performed under standard binding conditions. No robust or specific binding was observed under any condition tested. The protocol details are available upon request.
4.7. Structural Modeling Using AlphaFold3
Protein–protein interaction modeling between NsrR and RpoE was performed using AlphaFold3. Amino acid sequences of full-length E. coli NsrR and RpoE were used as input to the AlphaFold server with default options. Top5 models and experimental structures 6JBQ and 1OR7 in PDB were download and examined in UCSF ChimeraX [53] to generate the illustrations.
4.8. Statistical Analysis
All the statistical analyses were performed using GraphPad Prism 10. One-way ANOVA was used to assess group differences. Bartlett’s or Brown–Forsythe tests were applied to evaluate assumptions of equal variance, depending on the dataset. When comparing two groups, unpaired t-tests were used. Unless otherwise noted, the data represent the mean of at least three independent biological replicates. Bars and connected scatter points display group means; error bars represent the standard error of the mean (SEM). Specific statistical tests and significance thresholds are noted in individual figure legends.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Raina S. Missiakas D. Georgopoulos C. The rpo E gene encoding the sigma E (sigma 24) heat shock sigma factor of Escherichia coli EMBO J.1995141043105510.1002/j.1460-2075.1995.tb 07085.x 7889935 PMC 398177 · doi ↗ · pubmed ↗
- 2Mescas J. Rouviere P.E. Erickson J.W. Donohue T.J. Gross C.A. The activity of sigma E, an Escherichia coli heat-inducible sigma-factor, is modulated by experssion of out membrane proteins Genes Dev.1993726182628827624410.1101/gad.7.12b.2618 · doi ↗ · pubmed ↗
- 3Erickson J.W. A Gross C. Identification of the sigma E subunit of Escherichia coli RNA polymerase: A second alternate sigma factor involved in high-temperature gene expression Genes Dev.198931462147110.1101/gad.3.9.14622691330 · doi ↗ · pubmed ↗
- 4Missiakas D. Mayer M.P. Lemaire M. Georgopoulos C. Raina S. Modulation of the Escherichia coli s E (Rpo E) heat-shock transcription-factor activity by the Rse A, Rse B and Rse C proteins Mol. Microbiol.19972435537110.1046/j.1365-2958.1997.3601713.x 9159522 · doi ↗ · pubmed ↗
- 5De Las Penans A. Connolly L. Gross C.A. The s E-mediated response to extracytoplasmic stress in Escherichia coli is transduced by Rse A and Rse B, two negative regulators of s E Mol. Microbiol.19972437338510.1046/j.1365-2958.1997.3611718.x 9159523 · doi ↗ · pubmed ↗
- 6Alba B.M. Leeds J.A. Onufryk C. Lu C.Z. Gross C.A. Deg S and Yae L participate sequentially in the cleavage of Rse A to activate the ςE-dependent extracytoplasmic stress response Genes Dev.2002162156216810.1101/gad.100890212183369 PMC 186436 · doi ↗ · pubmed ↗
- 7Walsh N.P. Alba B.M. Bose B. A Gross C. Sauer R.T. OMP Peptide Signals Initiate the Envelope-Stress Response by Activating Deg S Protease via Relief of Inhibition Mediated by Its PDZ Domain Cell 2003113617110.1016/S 0092-8674(03)00203-412679035 · doi ↗ · pubmed ↗
- 8Snyder W.B. Davis L.J. Danese P.N. Cosma C.L. Silhavy T.J. Overproduction of Nlp E, a new outer membrane lipoprotein, suppresses the toxicity of periplasmic Lac Z by activation of the Cpx signal transduction pathway J. Bacteriol.19951774216422310.1128/jb.177.15.4216-4223.19957635808 PMC 177165 · doi ↗ · pubmed ↗