Early-Onset Retinal Dysfunction Associated with Novel WDR19 Variants in Sensenbrenner Syndrome
Bogumiła Wójcik-Niklewska, Zofia Oliwa, Zofia Zdort, Adrian Smędowski

TL;DR
A 2-year-old boy with Sensenbrenner syndrome showed early retinal dysfunction linked to new WDR19 gene variants, highlighting the need for early eye screening in this condition.
Contribution
The study reports two novel WDR19 variants and emphasizes early retinal dysfunction in Sensenbrenner syndrome.
Findings
Novel compound heterozygous WDR19 variants c.1778G>T and c.3536T>G were identified in a CED patient.
Early-onset retinal dysfunction was detected via severely reduced ERG responses and optic nerve hypoplasia.
The case underscores the importance of ophthalmologic screening for early detection in CED.
Abstract
Sensenbrenner syndrome, or cranioectodermal dysplasia (CED), is a rare autosomal recessive ciliopathy characterized by craniofacial, skeletal, ectodermal, and renal abnormalities. Ocular involvement, though infrequent, can include retinal dystrophy with early-onset visual impairment. We report a case of a 2-year-old boy with classic clinical features of CED and significant ocular findings. Genetic testing revealed two novel compound heterozygous variants in the WDR19 gene—c.1778G>T and c.3536T>G—expanding the known mutational spectrum associated with this condition. Ophthalmologic evaluation demonstrated bilateral optic nerve hypoplasia, high hyperopia, and severely reduced ERG responses, consistent with global retinal dysfunction. Fundoscopy revealed optic disk pallor, vessel attenuation, and peripheral pigment changes. Multisystem findings included postaxial polydactyly,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
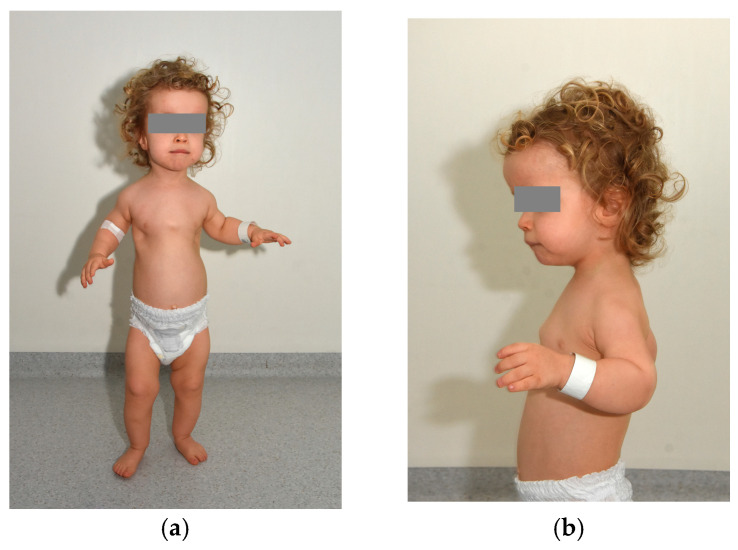 Figure 1
Figure 1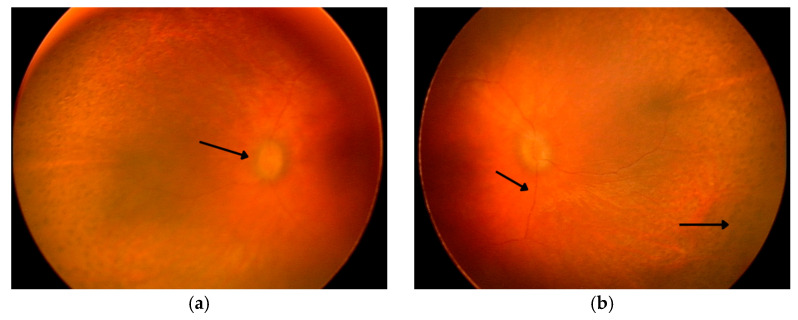 Figure 2
Figure 2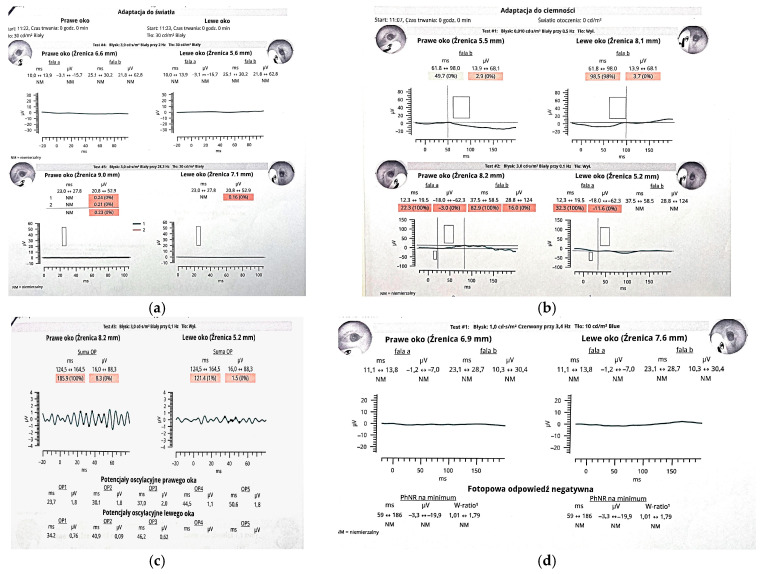 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and Kidney Cyst Diseases · Fetal and Pediatric Neurological Disorders · Cerebrospinal fluid and hydrocephalus
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Amar M.J.A. Sutphen R. Kousseff B.G. Expanded phenotype of cranioectodermal dysplasia (Sensenbrenner syndrome)Am. J. Med. Genet.19977034935210.1002/(SICI)1096-8628(19970627)70:4<349::AID-AJMG 3>3.0.CO;2-O 9182772 · doi ↗ · pubmed ↗
- 2Tan W. Lin A. Keppler-Noreuil K. Cranioectodermal Dysplasia. Gene Reviews® [Internet]Available online: https://www.ncbi.nlm.nih.gov/books/NBK 154653/(accessed on 29 June 2025)
- 3Bredrup C. Saunier S. Oud M.M. Fiskerstrand T. Hoischen A. Brackman D. Leh S.M. MidtbøM. Filhol E. Bole-Feysot C. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR 19Am. J. Hum. Genet.20118963464310.1016/j.ajhg.2011.10.00122019273 PMC 3213394 · doi ↗ · pubmed ↗
- 4Mukhopadhyay S. Wen X. Chih B. Nelson C.D. Lane W.S. Scales S.J. Jackson P.K. TULP 3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia Genes. Dev.2010242180219310.1101/gad.196621020889716 PMC 2947770 · doi ↗ · pubmed ↗
- 5Taschner M. Bhogaraju S. Lorentzen E. Architecture and function of IFT complex proteins in ciliogenesis Differentiation 201283 S 12S 2210.1016/j.diff.2011.11.00122118932 PMC 3977345 · doi ↗ · pubmed ↗
- 6Hildebrandt F. Benzing T. Katsanis N. Ciliopathies N. Engl. J. Med.20113641533154310.1056/NEJ Mra 101017221506742 PMC 3640822 · doi ↗ · pubmed ↗
- 7Yoshikawa T. Kamei K. Nagata H. Saida K. Sato M. Ogura M. Ito S. Miyazaki O. Urushihara M. Kondo S. Diversity of renal phenotypes in patients with WDR 19 mutations: Two case reports Nephrology 20172256657110.1111/nep.1299628621010 · doi ↗ · pubmed ↗