The first mitogenome of Petrovinema skrjabini from Equus ferus przewalskii: a phylogenetic analysis within the Strongylidae family
Huiping Jia, Liping Tang, Yajun Fu, Yu Xiong, Liping Yan, Changliang Shao, Kai Li, Dong Zhang, Defu Hu

TL;DR
This study reports the first complete mitochondrial genome of Petrovinema skrjabini, a parasitic nematode in horses, and uses it to clarify its evolutionary relationships within the Strongylidae family.
Contribution
The first complete mitogenome of Petrovinema skrjabini is sequenced and used to revise phylogenetic relationships in Strongylidae.
Findings
The P. skrjabini mitogenome is 13,885 base pairs with a high AT content of 75.4%.
Phylogenetic analysis suggests Triodontophorus should be reclassified into the subfamily Cyathostominae.
The study provides genetic markers for improved identification and taxonomy of P. skrjabini.
Abstract
Petrovinema skrjabini (Nematoda: Strongylidae, Cyathostominae) is a parasitic nematode colonizing the cecum and colon of equids. Like other cyathostomins, its larvae (L3) invade the intestinal mucosa, forming encysted nodules that may remain dormant for years. Mass larval emergence triggers larval cyathostominosis—a severe syndrome characterized by hemorrhagic typhlocolitis and diarrhea, with mortality rates exceeding 50%. However, owing to the morphological indistinguishability of cyathostomin and frequent mixed infections in natural settings, species-specific contributions to pathogenesis remain unresolved. Previous studies on P. skrjabini have predominantly focused on its morphology, with limited molecular information available. The complete mitogenome of Petrovinema skrjabini was sequenced using the Illumina NovaSeq 6000 platform, followed by assembly and annotation. We performed a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
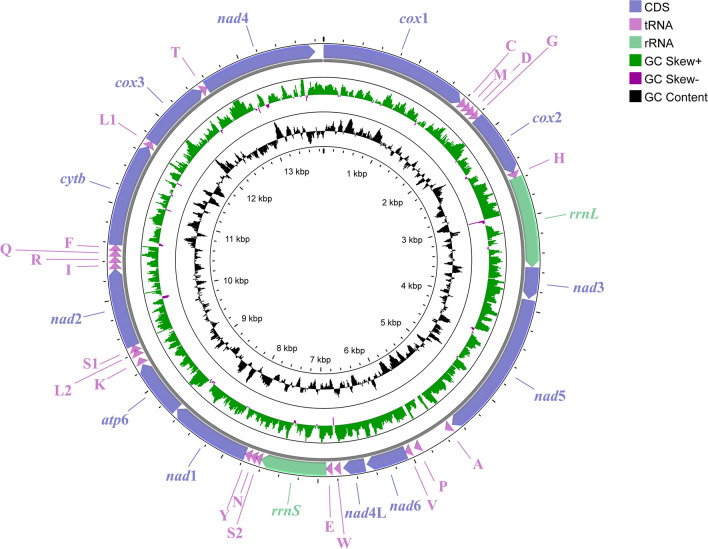 Figure 1
Figure 1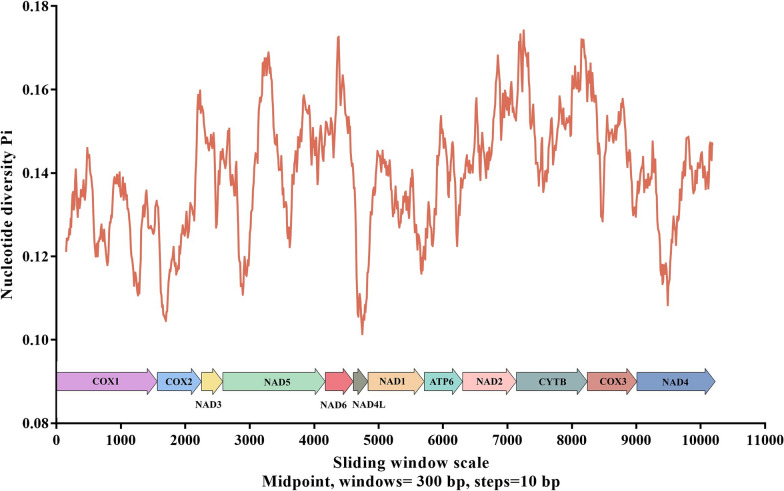 Figure 2
Figure 2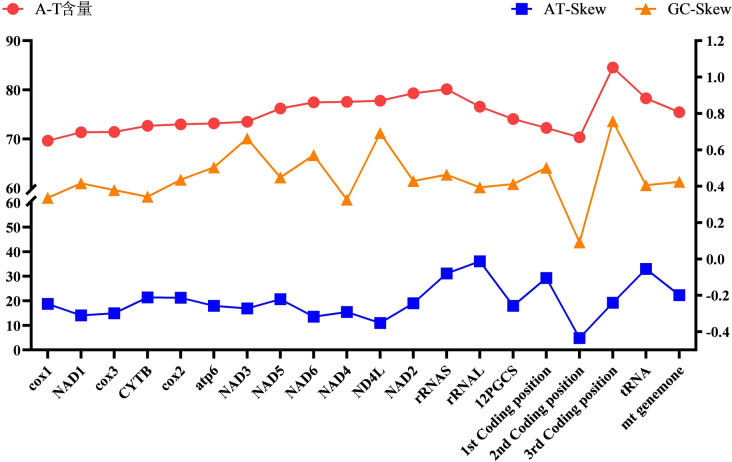 Figure 3
Figure 3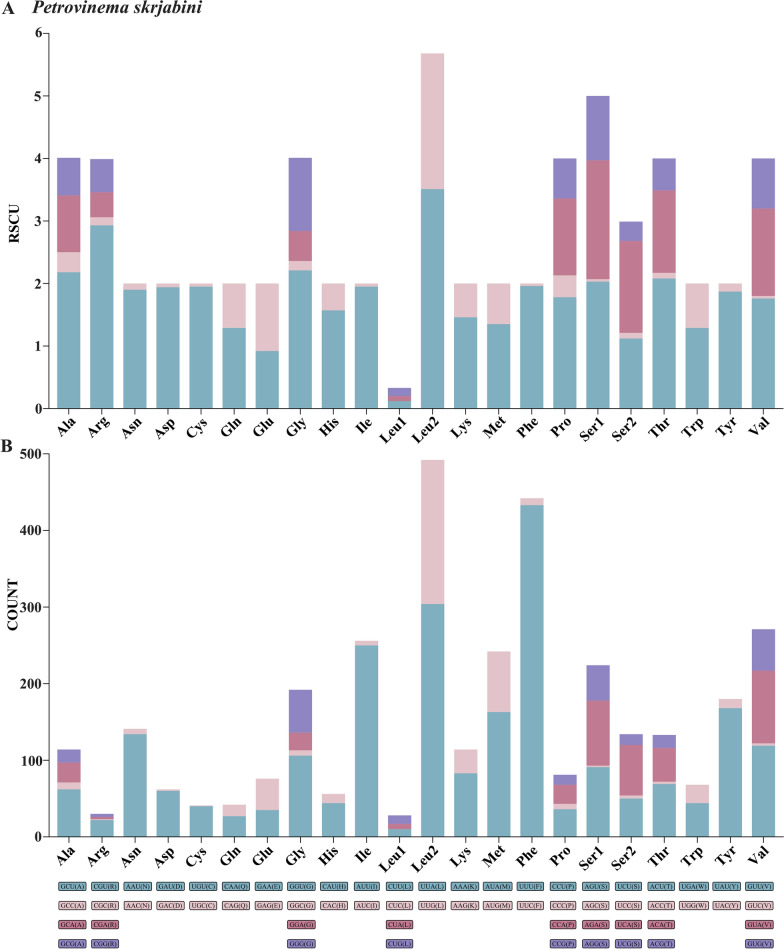 Figure 4
Figure 4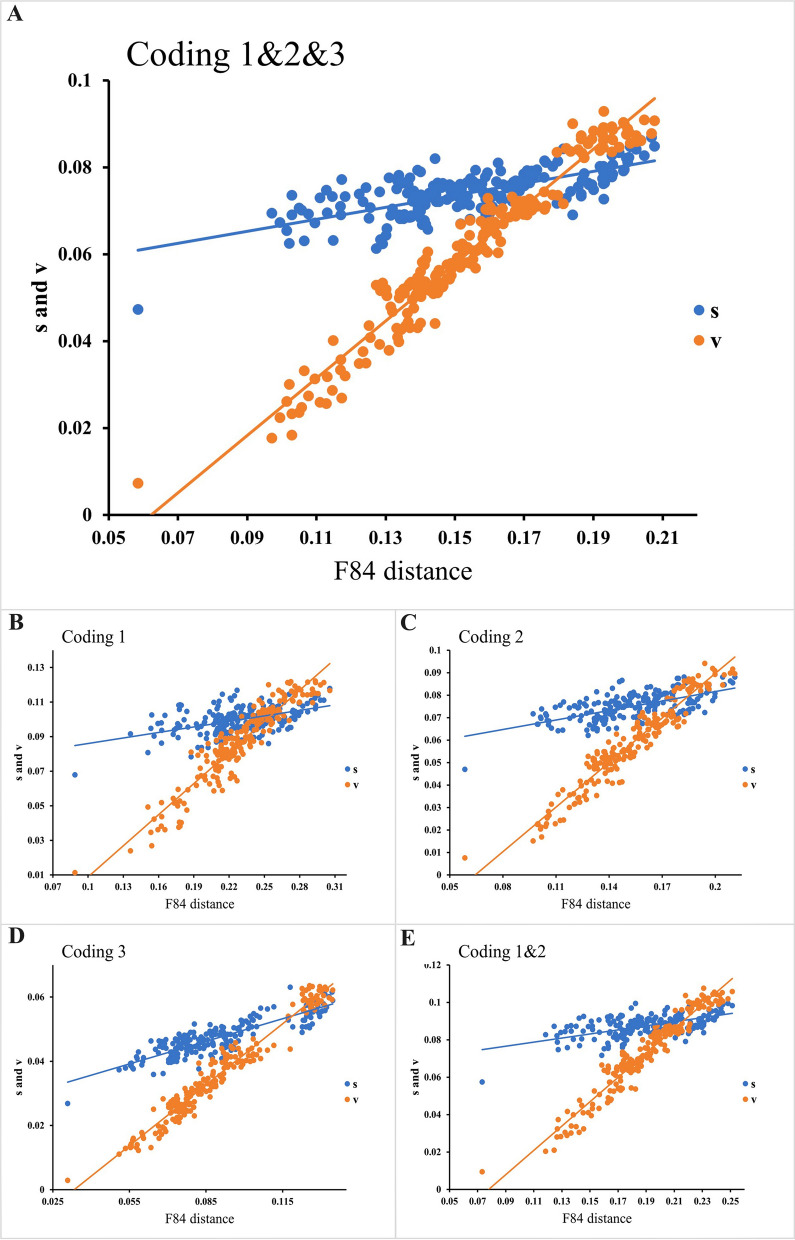 Figure 5
Figure 5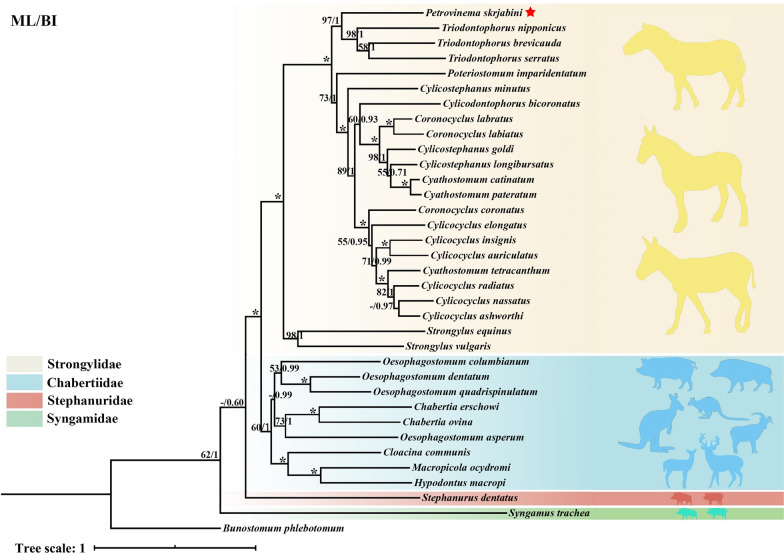 Figure 6
Figure 6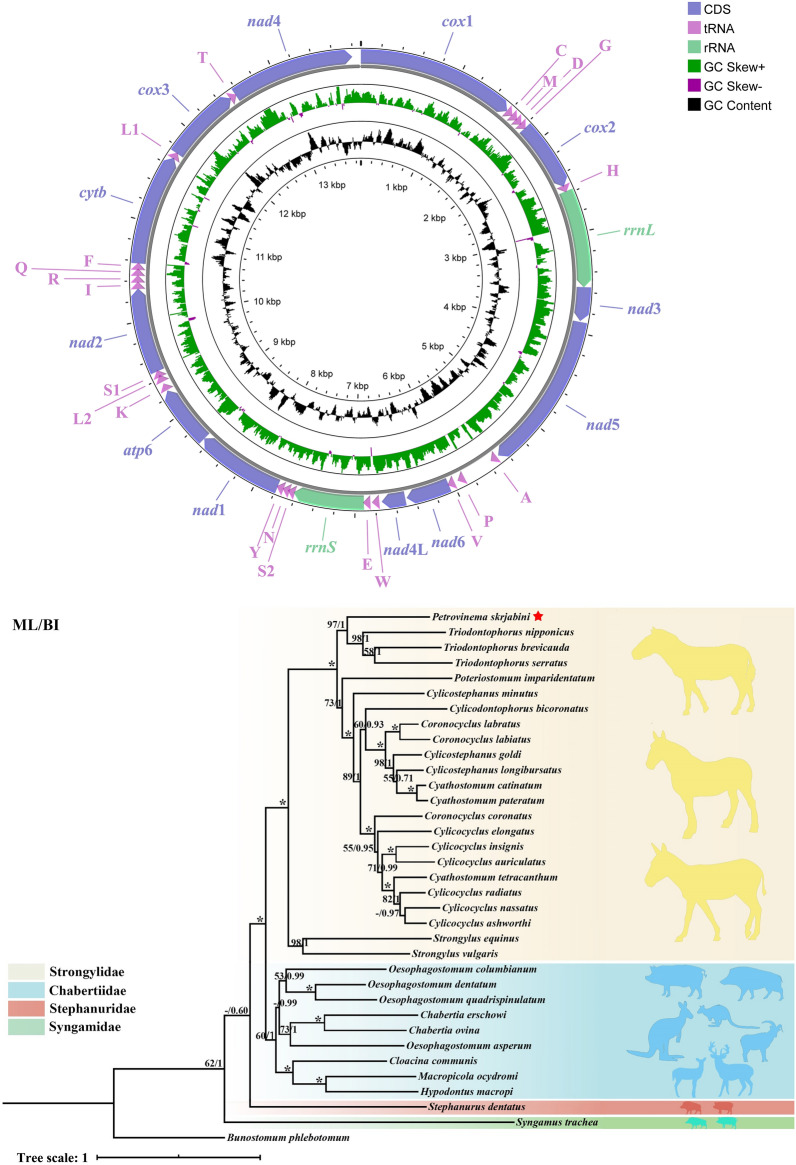 Figure 7
Figure 7- —The National Key R&D Program of China
- —The Beijing Forestry University Outstanding Young Talent Cultivation Project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHelminth infection and control · Genetic diversity and population structure · Mollusks and Parasites Studies
Background
Strongylids (Nematoda: Strongylidae) are a group of parasitic nematodes that co-infect the gastrointestinal tracts of equids [1–3], with veterinary and economic importance for domestic animals and wildlife [4–10]. Co-infecting nematode species can interact within the host, changing infection dynamics and transmission, with implications for host diseases [11–14]. Hence, a detailed understanding of strongylid species and how they infect the host is essential.
Studies on strongylid communities in equids rely heavily upon the morphological identification of specimens collected from feces or culled animals [15]. This method is challenging and labor-intensive, limiting the number of hosts that can be examined and requiring considerable expertise [15, 16]. Traditionally, Strongylidae is divided into two subfamilies, Strongylinae (large strongyles with globular or funnel-shaped buccal capsules) and Cyathostominae (small strongyles with cylindrical buccal capsules), based solely on morphological characteristics [22]. However, molecular studies are inconsistent with morphological classification criteria [19, 25–27]. For instance, Gao et al. showed through mitochondrial genome analyses that the genus Triodontophorus, morphologically assigned to Strongylinae, cluster within Cyathostominae in mitochondrial phylogenies [30], demonstrating the limitations of relying solely on buccal capsule morphology. These findings highlight the need for molecular approaches to reconcile taxonomic uncertainties. Advances in molecular technologies open promising avenues for nematode research, providing tools for species identification, exploring molecular evolution, and conducting phylogenetic studies [17–21]. To date, approximately 64 species from 19 genera in the Strongylidae family have been described [22]; however, only 22 compete mitogenomes belonging to 8 genera within Strongylidae are available in the GenBank Database (https://www.ncbi.nlm.gov/; accessed on 20 November 2023) [23, 24].
Petrovinema, described by Ershov in 1943, belongs to the phylum Nematoda, family Strongylidae, and subfamily Cyathostominae. It primarily infects equids such as donkeys and horses. Infection occurs via the fecal–oral route, causing symptoms such as subcutaneous edema, diarrhea, fever, and emaciation, with approximately 50% mortality [4, 28]. In 1975, Lichtenfels classified two species of Petrovinema, P. skrjabini and P. poculatum, into the genus Cylicostephanus. In 1986, Hartwich suggested that two species of Petrovinema transferred to the genus of Cylicostephanus is unreasonable based on morphological characteristics [22], which supported using the ITS 2 sequence by Hung et al. and recognized Petrovinema as an independent genus [23]. Currently two species of the genus Petrovinema are accepted: P. skrjabini and P. poculatum. In comparison with that of P. poculatum, the distribution range of P. skrjabini is relatively limited, has only been reported in Asia [22, 29], and lacks molecular data, which, to a certain extent, hinders the attainment of a comprehensive understanding of this strongylid species. Advanced studies of the genus Petrovinema have mainly focused on morphological evidences, while only a small number of studies have addressed their phylogenetic relationships based on molecular sequences, especially those including mitogenomes [22].
Therefore, this study addresses this critical gap by determining the complete mitogenome of P. skrjabini,, which will enable a more accurate identification of the species, and thus help to determine the specific parasite type of infection. Moreover, we sought to resolve taxonomic controversies related to the Strongylidae family.
Methods
Sample collection and DNA extraction
Petrovinema skrjabini specimens were collected from Equus ferus przewalskii dewormed with ivermectin in the Kalamaili Nature Reserve (Xinjiang, China). The specimens were thoroughly washed with saline solution, fixed in 70% ethanol, and stored at −40 °C. Nematodes were identified using a compound microscope (Olympus CX22, Japan), with reference to taxonomic keys and descriptions [22]. A tissue sample was taken from an adult P. skrjabini specimen for DNA extraction. The remaining body parts of the specimen were preserved as vouchers, labeled with the number 418–6, and then deposited in the Museum of Beijing Forestry University (Ne-418). DNA was extracted using the QIAamp DNA Blood Mini Kit, following the manufacturer’s instructions (Qiagen, GmbH, Germany), and eluted from the column using 200 μL of deionized water.
DNA annotation, sequence analysis, and comparative analysis
To survey the P. skrjabini genome, we generated TruSeq libraries using the Illumina NovaSeq 6000 platform (with 150 base pair [bp] paired-end reads) at Berry Genomics in China. Trimmomatic v0.38 [30] was used to eliminate highly redundant and low-quality sequences from the FASTQ data generated using the Illumina platform. De novo assemblies were created using IDBA-UD (http://i.cs.hku.hk/~alse/hkubrg/projects/idba_ud/) with a similarity threshold of 98% and k-values ranging from 80 to 124 bp considered. The COXI gene (GenBank: KP693427.1) sequences of close relatives (P. poculatum) served as “bait” references for obtaining the most appropriate and targeted mitochondrial scaffolds. The P. skrjabini mitogenome was annotated using the MITOS webserver [31], and mitochondrial DNA maps were generated utilizing the Proksee Server (https://proksee.ca/).
Protein-coding genes (PCGs) and ribosomal-RNA (rRNA) genes were compared with homologous genes from other strongylid nematodes using MEGA version 6 [32] (Table 1). The AT/GC skew, a metric for assessing strand bias, was computed as described by Perna and Kocher (1995) [33]. To analyze codon usage, the relative synonymous codon usage (RSCU) values of 12 PCGs (with their termination codons removed) were calculated using MEGA v6 [34]. The synonymous (Ks) and non-synonymous (Ka) substitution rates of the PCGs were computed using DnaSP version 5 [35]. We sorted the output data in terms of codon usage and counts and created graphics using the ggplot2 package implemented in R v3.4.1 [36]. Pairwise alignments of the 12 PCGs were conducted using MEGA v6 [32] to identify variable nucleotide sites. Sequence variability across the Strongylidae family, encompassing 22 species (including P. skrjabini), was assessed through sliding-window analysis using DnaSP v5 [37]. The sliding-window analysis was carried out as previously described [38]. To evaluate substitution saturation at each codon position in the PCGs, we employed Xia’s test [39] within the DAMBE.Table 1. Species used for reconstructing relationships in the present studySuperfamilyFamilySubfamilySpeciesLocusSequence length (bp)StrongyloideaStrongylidaeCyathostominaeCylicodontophorus bicoronatusNC_04214113,756Coronocyclus coronatusOR834861.113,870Coronocyclus labiatusNC_04223413,827Coronocyclus labratusMN83273613,856Cylicocyclus radiatusNC_03964313,836Cylicocyclus ashworthiNC_04671113,876Cylicocyclus auriculatusNC_04384913,831Cylicocyclus elongatusNC_06074813,875Cylicocyclus insignisNC_01380813,828Cylicocyclus nassatusNC_03229913,846Cyathostomum catinatumNC_03500313,838Cyathostomum pateratumNC_03807013,822Cyathostomum tetracanthumMN79280013,839Cylicostephanus goldiAP01768113,827Cylicostephanus longibursatusNC_081015.113,807Cylicostephanus minutusNC_03500413,826Poteriostomum imparidentatumNC_03500513,817Strongylus equinusNC_02686814,545Strongylus vulgarisGQ88871714,301Triodontophorus brevicaudaNC_02672914,305Triodontophorus nipponicusNC 03151713,701Triodontophorus serratusKX18515413,794ChabertiidaeChabertiinaeChabertia erschowiKF66060313,705Chabertia ovinaNC_01383113,682CloacininaeCloacina communisNC_06762613,689OesophagostominaeOesophagostomum asperumNC_02393213,672Oesophagostomum columbianumNC_02393313,561Oesophagostomum dentatumGQ88871613,869Oesophagostomum quadrispinulatumNC_01418113,681PhascolostrongylinaeHypodontus macropiNC_02309813,634Macropicola ocydromiNC_02309913,659Syngamidae–Syngamus tracheaNC_01382114,647Stephanuridae–Stephanurus dentatusMW97002913,735AncylostomatoideaAncylostomatidaeBunostominaeBunostomum phlebotomum (outgroup)NC_01230813,790
Phylogenetic analyses
Phylogenetic analyses were performed using 12 PCG matrixes from the mitogenome of P. skrjabini (this study) and 33 other Strongyloidea mitogenomes (obtained from the NCBI GenBank database; Table 1), with Bunostomum phlebotomum selected as the outgroup species [40]. Maximum likelihood (ML) and Bayesian inference (BI) analyses were performed using RAxML v8 [41] and MrBayes v3.2.7a [42] as described by Zhao et al. (2021, 2022) [43, 44]. The best substitution models for the 12 PCG sequences were determined using ModelTest-NG software v0.1.7 [45]. The general time-reversible (GTR) + invariable sites (I) + gamma distribution (G) substitution model was used for the ML analysis, involving 1000 bootstrap replications. For the BI analysis, two independent runs of four Markov chains (three heated, one cold) were conducted for two million generations, sampling every 1000 generations. The GTR + I + G substitution model was used. The first 25% of the sampled trees were discarded as burn-in. Convergence of the MCMC chains was assessed by monitoring the average standard deviation of split frequencies, which fell below 0.01. The phylogenetic trees were visualized using the iTOL online tool (https://itol.embl.de) [46].
Results
Composition and organization of the mitogenome
The complete mitogenome of P. skrjabini spanned 13,885 bp in length and contained 36 genes, including atp6, cox1–cox3, cytb, nad1–nad6, and nad4L, as well as the small and large subunit rRNA genes (rrnS and rrnL), 22 tRNA genes (tRNA-Ala, tRNA-Cys, and tRNA-Met, etc.) and two non-coding control regions (NCRs) (Fig. 1; Table 2), and has been deposited in the NCBI database (GenBank accession number: OP784481). All genes were encoded by the same strand, and the gene arrangement (GA) conformed to GA3 [47]. Notably, the P. skrjabini mitogenome contained 16 intergenic spacers, ranging from 1 to 54 bp in length, and five overlaps, reflecting a highly compact organization (Table 2). The overall AT content in the P. skrjabini mitogenome was 75.4%, with an AT skew of −0.20 and a CG skew of 0.42 (Fig. 2). Sliding-window analysis across the 12 PCGs revealed that nad4L had the lowest nucleotide variability, while cytb showed the highest (Fig. 3). Among the most conserved genes were nad4L, cox2, and nad4; whereas, cytb, cox3, and nad6 were the least conserved.Fig. 1. Organization of the complete mitochondrial genome of Petrovinema skrjabiniTable 2. Organization of the complete mitochondrial genome of Petrovinema skrjabiniGenes/regionsPositions and sequence lengths (bp)Intergenic nucleotidesInitiation/stop codonscox11–1578 (1578)0ATT/TAAtRNA-Cys(tgc)1585–1640 (56)6tRNA-Met(atg)1648–1706 (59)7tRNA-Asp(gac)1708–1765 (58)1tRNA-Gly(gga)1770–1824 (55)4cox21825–2520 (696)0ATT/TAAtRNA-His(cac)2520–2573 (54)−1rrnL2572–3547 (974)−2nad33548–3883 (336)0ATT/TAAnad53895–5478 (1584)11ATT/TAAtRNA-Ala(gca)5508–5562 (55)29LNCR5563–5889 (326)0tRNA-Pro(cca)5890–5943 (54)0tRNA-Ser(gta)5998–6051 (54)54nad66052–6486 (435)0ATT/TAGnad4L6503–6736 (234)33ATT/TAGtRNA-Trp(tga)6770–6826 (57)31tRNA-Glu(gaa)6858–6915 (58)−1rrnS6915–7618 (704)−1tRNA-Ser2(tca)7618–7670 (53)0tRNA-Asn(aac)7671–7727 (57)7tRNA-Tyr(tac)7735–7789 (55)0nad17790–8662 (873)1TTG/TAGatp68664–9263 (600)12ATT/TAAtRNA-Lys(aaa)9276–9338 (63)34tRNA-Leu2(tta)9373–9427 (55)0tRNA-Ser1(aga)9428–9479 (52)0nad29480–10325 (846)2TTG/TAGtRNA-Ile(atc)10,328–10,387 (60)6tRNA-Arg(cgt)10,394–10,449 (56)11tRNA-Gln(caa)10,461–10,515 (55)3tRNA-Phe(ttc)10,519–10,575 (57)0cytb10,576–11688 (1113)−1ATT/TAAtRNA-Leu1(cta)11,688–11,743 (56)0cox311,744–12,509 (766)0ATT/TtRNA-Thr(aca)12,510–12,568 (59)0nad412,569–13,798 (1230)0TTG/TAASNCR13,799–13,885 (87)0Fig. 2A + T content and nucleotide skew of genes, individual elements, and the complete mitogenome of Petrovinema skrjabiniFig. 3. Sliding window analysis (window size 300 bp, step size 10 bp) of the alignment of mitochondrial protein-coding sequences of Petrovinema skrjabini used to estimate nucleotide diversity Pi (π) across the alignments. Nucleotide diversity was plotted against the mid-point positions of each window. Gene boundaries are indicated above the graph
PCGs and codon usage
A total length of 10,291 bp was assembled across the 12 PCGs of P. skrjabini, encoding 3419 amino acids (Table 3). The results showed that T was the most prevalent base in all 12 PCGs of P. skrjabini, followed by G, A, and C. The AT contents of the 12 PCGs ranged from 69.65% (cox1) to 79.31% (nad2), with the third codon positions exhibiting the highest AT content (84.55%) compared with the first (72.25%) and second positions (70.35%). The high usage frequencies of codons such as UUU (Phe) and UUA (Leu) contributed to the overall high AT content of the mitogenome, whereas codons such as CUC (Leu) and GUC (Val) were rarely used (Fig. 4A and B).Table 3. Lengths and amino acids of mitochondrial genes/regions of 17 Strongylidae speciesPet. skrjabiniCya. catinatumCya. pateratumCya. tetracanthumCylicos. goldiCylicos. minutusCor. labiatusCor. labratusCylicoc. auriculatusCylicoc. insignisCylicoc. elongatusCylicoc. nassatusCylicoc. radiatusCylicoc. ashworthiCylicod. bicoronatusPot. imparidentatumStr. equinusStr. vulgarisTri. brevicaudaTri. nipponicusTri. serratuscox11578/5251578/5251575/5241578/5251578/5251578/5251578/5251578/5251578/5251578/5251578/5251578/5251578/5251578/5251578/5251578/5251578/5251578/5251578/5251578/5251578/525cox2696/231696/231696/231696/231696/231696/231696/231696/231696/231696/231696/231696/231696/231696/231696/231696/231696/231697/231696/231696/231696/231rrnL974/–976/–976/–977/–972/–972/–979/–975/–969/–959/–968/–974/–969/–978/–982/–983/–959/–959/–975/–976/–961/–nad3336/111336/111336/111336/111336/111336/111336/111336/111336/111336/111336/111336/111336/111336/111336/111336/111336/111336/111336/111336/111336/111nad51584/5271584/5271584/5271584/5271593/5301584/5271584/5271584/5271584/5271584/5271584/5271584/5271584/5271584/5271584/5271584/5271599/5321584/5271584/5271584/5271584/527nad6435/144435/144435/144435/144435/144435/144435/144435/144435/144435/144435/144435/144435/144435/144438/145435/144435/144435/144435/144435/144435/144nad4L234/77234/77234/77234/77234/77234/77234/77234/77234/77234/77234/77234/77234/77234/77234/77234/77234/77234/77234/77234/77234/77rrnS704/–708/–698/–700/–699/–700/–701/–701/–699/–700/–699/–699/–697/–711/–702/–709/–708/–700/–703/–696/–701nad1873/290873/290873/290873/290873/290873/290873/290873/290873/290873/290873/290873/290873/290873/290873/290873/290879/292876/291873/290873/290873/290atp6600/199600/199600/199600/199600/199600/199600/199600/199600/199600/199600/199600/199600/199600/199600/199600/199600/199603/200600/199600/199600/199nad2846/281846/281846/281846/281846/281846/281846/281846/281846/281846/281846/281846/281846/281846/281846/281846/281846/281846/281846/281846/281846/281cytb1113/3701113/3701113/3701113/3701113/3701113/3701110/3691113/3701113/3701113/3701113/3701113/3701113/3701113/3701113/3701113/3701113/3701116/3711113/3701113/3701113/370cox3766/255766/255766/255766/255766/255766/255766/255766/255766/255769/256766/255766/255766/255766/255766/255766/255766/255766/255766/255766/255766/255nad41230/4091230/4091230/4091230/4091230/4091230/4091230/4091230/4091230/4091230/4091230/4091230/4091230/4091230/4091230/4091227/4081230/4091230/4091230/4091230/4091230/409Total11,96711,97511,96211,96811,97111,96311,96811,96711,95911,95311,95811,96411,95711,98011,97811,98011,97911,96011,96911,96311,953Pet. skrjabini, Petrovinema skrjabini; Cya. Catinatum, Cyathostomum catinatum; Cya. pateratum, Cyathostomum pateratum; Cya. Tetracanthum, Cyathostomum tetracanthum; Cylicos. goldi, Cylicostephanus goldi; Cylicos. minutus, Cylicostephanus minutus; Cor. labiatus, Coronocyclus labiatus; Cor. labratus, Coronocyclus labratus; Cylicoc. auriculatus, Cylicocyclus auriculatus; Cylicoc. insignis, Cylicocyclus insignis; Cylicoc. elongatus, Cylicocyclus elongatus; Cylicoc. nassatus, Cylicocyclus nassatus; Cylicoc. radiatus, Cylicocyclus radiatus; Cylicoc. ashworthi, Cylicocyclus ashworthi; Cylicod. bicoronatus, Cylicodontophorus bicoronatus; Pot. imparidentatum, Poteriostomum imparidentatum; Str. equinus, Strongylus equinus; Str. vulgaris, Strongylus vulgaris; Tri. brevicauda, Triodontophorus brevicauda; Tri. nipponicus, Triodontophorus nipponicus; Tri. serratus, Triodontophorus serratusFig. 4. Relative synonymous codon usage (RSCU, A) and COUNT (B) of Petrovinema skrjabini mitogenome
tRNA genes, rRNA genes, and non-coding regions
The P. skrjabini mitogenome contained 22 tRNA genes, ranging in length from 52 to 63 bp, these tRNAs appeared to fold into an atypical clover-leaf secondary structure. Two tRNAs, namely tRNA-Ser2 and tRNA-Ser1, lacked a dihydrouridine arm and had a d-loop structure. The other 20 tRNAs lacked the TΨC loop, which was replaced by the “TV-replacement loop”. The two rRNA genes, rrnL (974 bp) and rrnS (704 bp), were located between tRNA-His and nad3, and between tRNA-Glu and tRNA-Ser, respectively. The AT contents of rrnL and rrnS were 80.1% and 76.6%, respectively. Two NCRs were identified: the long NCR (326 bp) located between tRNA-Ala and tRNA-Pro, and the short NCR (87 bp) located between nad4 and cox1 (Table 2), with AT contents of 87.8% and 80.5%, respectively. The rrnL gene was located between tRNA-His and nad3, whereas rrnS was located between tRNA-Glu and tRNA-Ser.
Phylogenetic analyses
Prior to phylogenetic reconstruction, substitution saturation analysis of the 12 PCGs was performed (Fig. 5). The substitution saturation index (Iss) was significantly lower than the critical Iss value (P < 0.001), confirming minimal saturation and validating the suitability of these sequences for phylogenetic inference. Phylogenetic analyses were reconstructed using concatenated sequences of the 12 PCGs from P. skrjabini and 33 other Strongyloidea species representing 16 genera (data from GenBank). The maximum likelihood (ML) and Bayesian inference (BI) analyses produced identical tree topologies (Fig. 6). While most nodes were well-supported, some nodes in Oesophagostominae and Strongylidae displayed weak bootstrap support (< 50) in ML but were strongly supported in BI (posterior probability ≥ 0.97). The Cyathostominae subfamily formed a strongly supported monophyletic clade (bootstrap = 100; posterior probability = 1), including genera Cylicocyclus, Cyathostomum, Coronocyclus, Poteriostomum, Cylicostephanus, Cylicodontophorus, Petrovinema, and Triodontophorus (traditionally classified under Strongylinae). In contrast, Strongylinae was paraphyletic, with Strongylus vulgaris and S. equinus forming a basal clade, while Triodontophorus clustered within Cyathostominae. Petrovinema skrjabini clustered robustly with three species of the Triodontophorus genus (posterior probability = 1; bootstrap = 97), forming a monophyletic group that further clustered with six other genera within the Cyathostominae subfamily (Fig. 6).Fig. 5. The transitions (s) and transversions (v) for 12 protein-coding genes of Strongyloidea against GTR distance. Plots in blue and orange indicate transition and transversion, respectively. (A) Coding genes 1, 2 and 3; (B) Coding gene 1; (C) Coding gene 2; (D) Coding gene 3; (E) Coding genes 1 and 2Fig. 6Phylogenetic relationships of Strongyloidea inferred from mitochondrial genomes. Topology obtained on the basis of concatenated amino acid sequences of 12 protein coding genes analyzed by maximum likelihood (ML) and Bayesian inference (BI) using Bunostomum phlebotomum as an outgroup. Statistical support values (bootstrap/posterior probability) of ML/BI analysis are shown above the nodes. Asterisks (*) indicate ML/BI = 100/1.0, other values are given above the nodes. Families are highlighted by individual colors. The species identified in the present study is denoted by the red star. Values below 50% are shown here as “–”
Discussion
Previous studies on the Strongyloidea superfamily have primarily relied on single-gene markers or morphological characteristics, which often lack the resolution required to resolve complex evolutionary relationships, particularly for the genus Petrovinema. In this study, we sequenced and analyzed the complete mitochondrial genome of P. skrjabini, revealing its gene content, arrangement, and codon usage. This work not only expands the available genomic resources for Strongyloidea nematodes but also offers new insights into their evolutionary relationships and phylogenetic placement.
The gene arrangement of P. skrjabini conformed to the GA3 [47] pattern, consistent with other strongylids such as T. brevicauda, Strongylus equinus, Cylicocyclus insigne, and Cylicodontophorus bicoronatus [23, 24, 48–52]. This structural conservation reflects evolutionary stability and strong selective pressures within the Strongyloidea superfamily. The absence of the ATP8 gene in P. skrjabini and other Strongyloidea species, is likely due to functional redundancy or gene loss during evolution. The notable AT bias in the P. skrjabini mitogenome (75.4%) was consistent with Strongyloidea mitogenomes [24, 52–54] (Fig. 2), with NCR AT content (86.5%) similar to those observed in nematodes and trematodes, such as Cylicocyclus radiatus, Cyathostomum catinatum, Coronocyclus labiatus, Echinostoma miyagawai, and Postharmostomum commutatum [24, 55, 56]. In addition, the size and location of NCRs in the P. skrjabini mitogenome resembled those in the subfamily of Cyathostominae nematodes (Table 3) but differed from those of the subfamily of Strongylinae nematodes [48–50]. Regarding nucleotide composition, the highest AT-skew value was found at the third codon position, and the lowest in the second codon position, consistent with observations in nematodes and trematodes, such as Cylicocyclus radiatus, Coronocyclus labiatus, Cylicodontophorus bicoronatus, and E. miyagawai [23, 24, 55]. These findings suggest that the nucleotide composition and skewness in the P. skrjabini mitogenome are consistent with evolutionary patterns observed in related species, indicating conserved mutational pressures and selection mechanisms across different nematode and trematode taxa.
The mitochondrial codon usage in P. skrjabini and related Strongylidae species follows the standard invertebrate genetic code, with UUA (Leu) and CGU (Arg) being the most common, consistent with 22 published Strongylidae mitogenomes [23, 24, 50, 55]. Minor interspecific differences in RSCU values were observed: CUC (Leu) has an RSCU of 0.01 in Cylicodontophorus bicoronatus but was absent (RSCU = 0) in P. skrjabini and Coronocyclus labiatus. Similarly, CGC (Arg) showed limited usage (RSCU = 0.13) in P. skrjabini but was absent (RSCU = 0) in Cylicocyclus radiatus and Cylicodontophorus bicoronatus [23, 24]. These RSCU differences highlight evolutionary divergence in codon preference, not alterations to the genetic code, aligning with the conserved framework reported for Strongylidae nematodes [23, 24, 50, 52]. The rRNA genes, rrnL and rrnS, in P. skrjabini are of similar length to those in other nematodes, such as T. brevicauda (975 bp and 703 bp), Po. imparidentatum, Chabertia erschowi (970 bp and 696 bp), and Chabertia ovina (962 bp and 696 bp) [24, 34, 48, 50, 57, 58]. There was no difference compared with other Strongylinae nematodes, such as Cylicocyclus radiates [57], in the position of rrnS. The rrnL gene was located between tRNA-His and nad3; whereas, rrnS was located between tRNA-Glu and tRNA-Ser.
This study revealed the phylogenetic relationships of the genus Petrovinema through mitogenome analysis of P. skrjabini. Contrary to the findings of Bu et al. (2016), based on P. poculatum ITS1/ITS2 sequence data [59], which suggested a close phylogenetic relationship between Petrovinema and Poteriostomum, our mitochondrial genomic data demonstrate that P. skrjabini forms a monophyletic clade with Triodontophorus (bootstrap/posterior probability = 97/1). This difference could potentially be due to differences in gene fragment selection and length used for phylogenetic tree construction, as longer mitochondrial genome segments provide better resolution for evolutionary trees. Owing to its relatively long length, the mitochondrial genome contains more informative data and more accurately represents the evolutionary relationships among parasitic nematodes. Similarly, the taxonomic discordance between the mitochondrial clade of Petrovinema and the nuclear gene clade of Poteriostomum underscores the necessity of integrating multi-genomic datasets in resolving phylogenies of morphologically convergent groups.
The mitochondrial phylogenetic position of the genus Triodontophorus (nested within the subfamily Cyathostominae), exhibits a clear inconsistency with its morphological classification under Strongylinae (Fig. 6). This incongruence corroborates the conclusions of Hung et al. and Gao et al. [27, 60]. Our results further validate that the traditional classification, dividing Strongylidae into the Strongylinae and Cyathostominae subfamilies [22] based solely on the size and shape of the buccal capsule is insufficient. On the basis of the existing molecular data, we propose transferring Triodontophorus to Cyathostominae while retaining Strongylinae as a monotypic subfamily containing the genus Strongylus. The phylogenetic analysis in this study was limited by the insufficient coverage of genomic data for extant genera in the Strongylinae and Cyathostominae subfamilies (9/19 genera lacking data). Future studies should prioritize filling the genomic data for key genera (such as Oesophagodontus and Skrjabinodentus) to enhance the reliability of the topological structure. In addition, the coordinated analysis of morphological trait quantification (such as buccal capsule depth) and molecular evolutionary rates remains unexplored. Subsequent studies can further dissect the mechanisms of species differentiation by integrating multi-omics data from geographical population samples. These expansion directions will systematically improve the classification framework of the Strongylidae family and provide new dimensions for the study of the adaptive evolution of parasitic nematodes.
Conclusions
In the present study, the complete mitogenome sequence of P. skrjabini was assembled, and comparative mitogenomes and phylogenetic analyses with other Strongyloidae species were carried out. Using next-generation sequencing technology, our analyses provided additional molecular evidence to clarify the systematic position of P. skrjabini, further improving the understanding of phylogenetic relationships within the Strongylidae family. Our study enriches the database of Strongylidae mitogenomes. The availability of the complete mitogenome of P. skrjabini also provides useful genetic markers for molecular epidemiology, population genetics, and systematics of Strongylidae nematodes infecting equids.