Gene regulation in Cryptosporidium: New insights and unanswered questions
Samantha Gunasekera, Jessica C. Kissinger

TL;DR
This review explores how genes are regulated in the parasite Cryptosporidium, highlighting what is known and what remains unclear.
Contribution
The paper identifies two unique features of Cryptosporidium gene regulation: retention of E2F/DP1 and polycistronic transcription.
Findings
Most gene regulatory components in Cryptosporidium lack experimental validation.
Cryptosporidium retains the E2F/DP1 transcription factor family, a trait unique among apicomplexans.
C. parvum produces polycistronic transcripts, a rare feature in eukaryotes.
Abstract
Parasites of the genus Cryptosporidium have evolved to have a highly compact genome of ∼9.1 Mb. The mechanisms that regulate gene expression in Cryptosporidium spp. remain incompletely understood at all levels, including chromatin accessibility, transcription factor activation and repression and RNA processing. This review discusses possible mechanisms of gene regulation in Cryptosporidium spp., including histone modifications, cis regulatory elements, transcription factors and non-coding RNAs. Cryptosporidium spp. are among the most basal branching apicomplexans and existing evidence suggests that they diverge from other members of their phylum via retention of the E2F/DP1 transcription factor family, and the recent discovery that C. parvum produces polycistronic transcripts. Most of what we know about gene regulation in the genus Cryptosporidium is based on sequence conservation and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
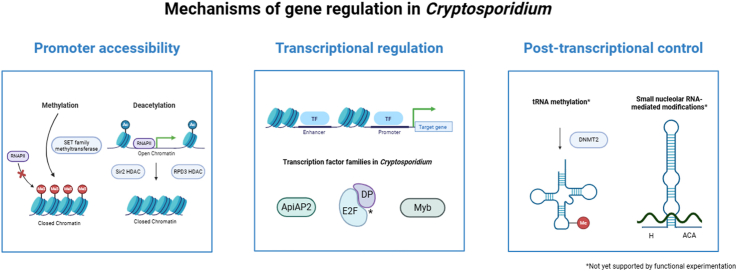 Figure 1
Figure 1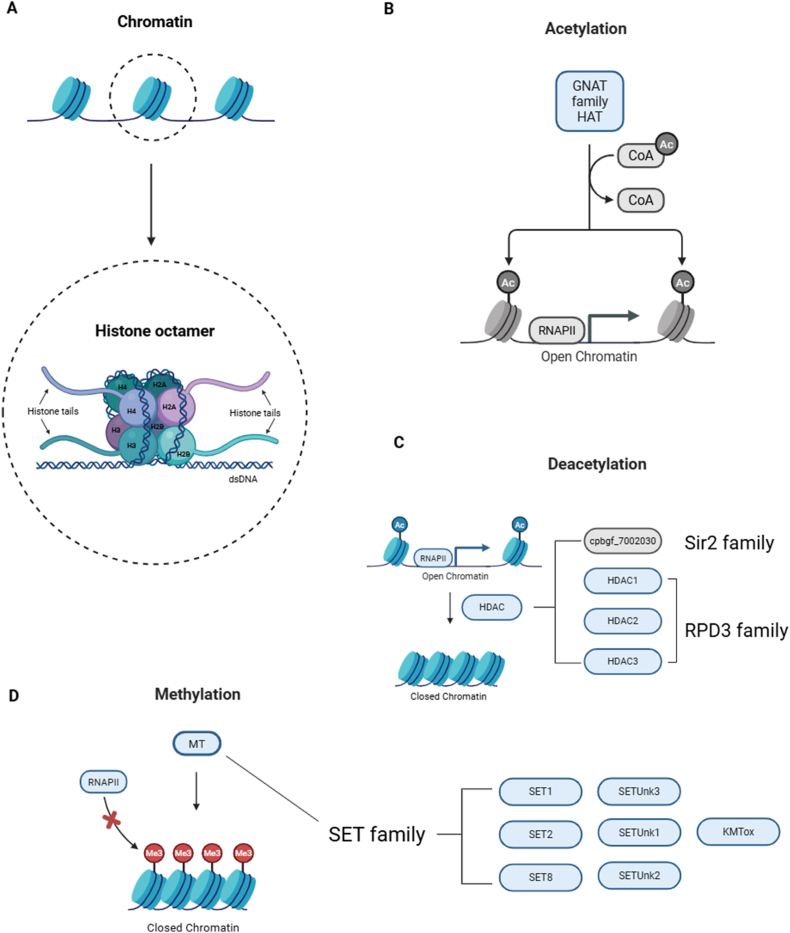 Figure 2
Figure 2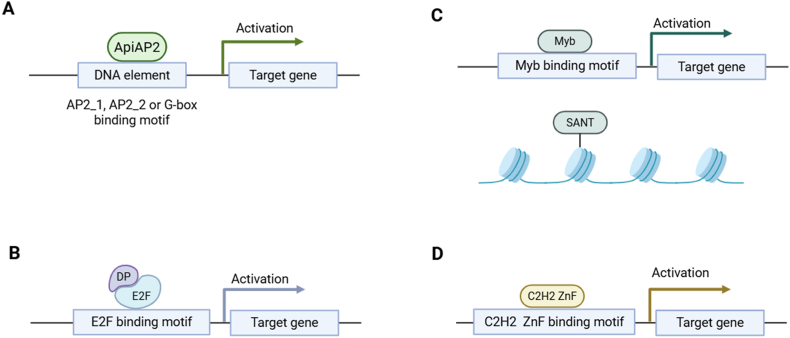 Figure 3
Figure 3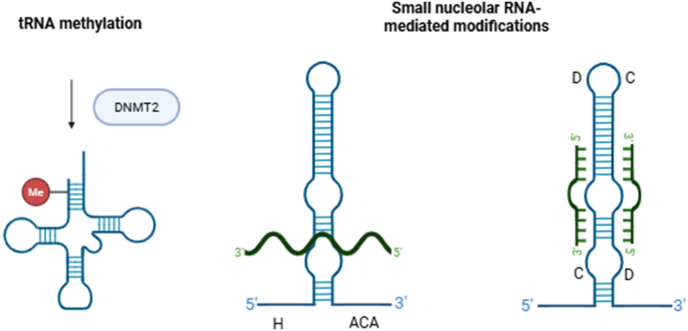 Figure 4
Figure 4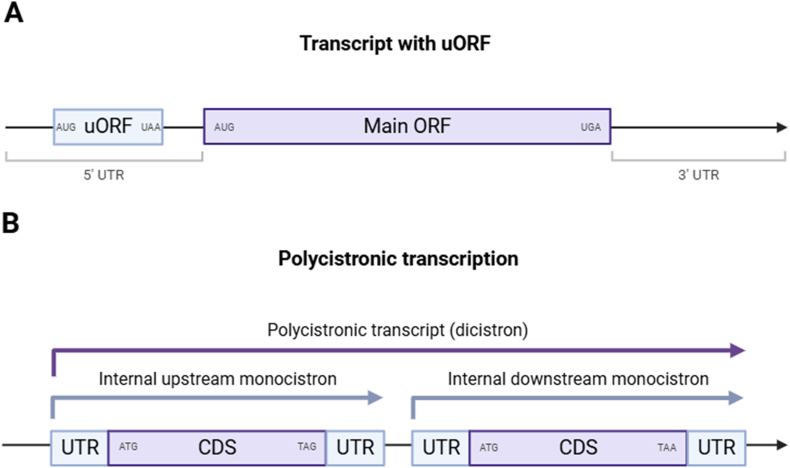 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsParasitic Infections and Diagnostics · Toxoplasma gondii Research Studies · Cytomegalovirus and herpesvirus research
Introduction
1
The transcriptional regulatory networks that control gene expression in Cryptosporidium spp. remain incompletely understood. Gene expression in Cryptosporidium spp. occurs in a tightly programmed cascade over the course of intracellular development, with distinct clusters of transcripts that rise and fall in abundance together as the parasite transitions through morphologically and functionally distinct life cycle stages (Mauzy et al., 2012; Oberstaller et al., 2013; Walzer et al., 2024). The prevailing dogma of apicomplexan gene regulation indicates that the production of transcripts occurs “just-in-time”, only if and when induction of the target gene is required (Bozdech et al., 2003). Synchronous gene expression patterns between co-regulated clusters of genes do not correlate with chromosomal location, with co-localized gene families often having vastly different expression profiles at different stages of the life cycle (Mauzy et al., 2012).
The genus Cryptosporidium is among the most basal-branching members of the phylum Apicomplexa and may provide insights into the ancestral mechanisms of gene regulation (Waller and Carruthers, 2024). Broadly, apicomplexans are well-documented to utilize an expansion of the apicomplexan AP2 (ApiAP2) family of transcription factors for gene regulation (Balaji et al., 2005), in the absence of many canonical eukaryotic transcription factors (Templeton et al., 2004). Cryptosporidium spp. are the only members of the phylum to have retained the E2F/DP1 transcription factor family, and is demonstrably less reliant on the ApiAP2 superfamily for gene regulation (Oberstaller et al., 2013).
In eukaryotes, the accessibility of cis regulatory elements by DNA binding proteins is controlled by chromatin structure, which is highly dynamic and regulated by a suite of histone-modifying enzymes and ncRNAs. Additionally, while most eukaryotes encode just one gene per mRNA molecule, polycistronic transcripts have been recently discovered in Cryptosporidium parvum sporozoites and merozoites (Xiao et al., 2025); a first for the Apicomplexa.
In this review, we will examine existing lines of scientific inquiry suggesting that Cryptosporidium parvum utilizes transcriptional regulatory systems that diverge somewhat from the model of gene regulation in other apicomplexans. We aim to provide an overview of what is already known about gene regulation in Cryptosporidium spp., and what remains to be elucidated.
Epigenetic regulation of gene expression
2
Histone tail post-translational modifications
2.1
In eukaryotes, nucleosomes are the fundamental organizational unit of chromatin. Each nucleosome is comprised of a histone octamer containing two copies each of the H2A, H2B, H3, and H4 histone core proteins wrapped with ∼147 bp of DNA (Fig. 1A) and is separated by 10–90 bp of linker DNA often bound by linker histones (Richmond and Davey, 2003). Nucleosomes are intrinsically dynamic, and function to regulate access to DNA by other nuclear factors. The presence of nucleosomes predates the divergence of eukaryotes and archaea in evolution, indicating that the role of chromatin architecture in gene regulation is ancient (Ammar et al., 2012). Linker histones are non-essential in lower organisms, and the C. parvum reference genome assembly has no annotated linker histone protein encoding genes (Abrahamsen et al., 2004).Fig. 1. Enzymes in C. parvum with possible roles in post-translational modifications of histone tails. A Schematic diagram of chromatin and histone octamer structure. BCryptosporidium parvum does possess GNAT family HAT orthologs but the HAT pathway has been shaded grey since none have been functionally confirmed. CCryptosporidium parvum encodes three RPD3 family HDACs which have been demonstrated to bind histone tails, and one Sir2 family HDAC that has not been experimentally confirmed (shown in grey). D Seven out of eight SET family proteins in C. parvum were identified as putative histone methyltransferases (Sawant et al., 2022). Created in Biorender. Gunasekera, S. (2025) https://BioRender.com/wp3u43V.Fig. 1
Histone core proteins each have an N-terminal histone tail domain and a C-terminal histone fold domain, though H2A monomers each possess an additional C-terminal histone tail. The histone tails are positively charged, facilitating the association with negatively charged DNA (Sterner and Berger, 2000). Histone tails are rich in lysine and arginine residues that are often the site of post-translational modifications, which can include acetylation, phosphorylation, methylation, ubiquitination, ADP-ribosylation, biotinylation, and lactylation, though this list is not exhaustive (Hansen, 2002). Different combinations of post-translational modifications mark functional units of chromatin that recruit proteins that can activate or repress gene expression, regulate DNA replication and chromatin remodelling. These post-translational modifications are collectively referred to as the histone code (Strahl and Allis, 2000). Epigenetic regulation of gene expression in Cryptosporidium spp. is under-studied, and only a subset of post-translational modifications of histone tails have been investigated.
Acetylation and deacetylation
2.1.1
Histone acetyltransferases (HATs) and deacetylases (HDACs) play key roles in transcriptional regulation (Strahl and Allis, 2000). HATs transfer an acetyl group from acetyl-CoA to a lysine residue in a histone tail, partially neutralizing its positive charge and weakening the histone-DNA association. The outcome is increased accessibility of a locus to transcriptional activation machinery. The removal of the acetyl group catalyzed by HDACs is associated with transcription repression. HATs and HDACs work synergistically to regulate transcription (Sterner and Berger, 2000).
Most HATs belong to the ancient GNAT superfamily and the MYST family (Iyer et al., 2008). The C. parvum reference genome has several annotated GNAT family acetyltransferases (Abrahamsen et al., 2004), though none have been the subject of functional investigation (Fig. 1A). No MYST family HATs are annotated in the C. parvum reference genome (Abrahamsen et al., 2004), though MYST protein domains are present in more recent Cryptosporidium spp. genome assemblies (Baptista et al., 2022). The HDACs are comparatively better studied in Cryptosporidium spp. (Fig. 1B) and can be divided into three structurally unrelated groups, the HD2 family, Sir2 family, and the RPD3 family. The HD2 family is unique to plants, while the Sir2 and RPD3 are superfamilies present across eukaryotes, prokaryotes and archaea (Iyer et al., 2008; Rider and Zhu, 2009). The RPD3 superfamily uses metal-dependent catalysis, and the Sir2 superfamily uses a NAD cofactor (Iyer et al., 2008). Cryptosporidium parvum possesses three RPD3 family HDAC proteins (Table 1) that are postulated to play a role in regulation of DNA replication (Rider and Zhu, 2009), and one putative Sir2 family protein (cpbgf_7002030/cgd7_2030) that has limited C. parvum-specific functional information available (Yasukawa and Yagita, 2010).Table 1RPD3 family HDACs in Cryptosporidium parvum.Table 1. Gene IDProtein nameaPredicted targetaLife cycle stagebSingle-cell atlas designationbcpbgf_60080/cgd6_80HDAC1–AsexualCluster 4cpbgf_6001380/cgd6_1380HDAC2–Not stage-specificClusters 3–5, 14, 15cpbgf_800480/cgd8_480HDAC3H4K8, H4K12Not stage-specificClusters 2, 6, 7, 10, 11, 14–17aThe RP3 family HDACs in C. parvum were originally characterized by Rider and Zhu (2009).bSingle-cell transcriptome atlas designation and life cycle stage(s) of expression from Walzer et al. (2024). Clusters 1–9 correspond to asexual development, 10–12 correspond to male sexual development, and 13–18 correspond to female sexual development.
Methylation and demethylation
2.1.2
Histone methyltransferases add methyl groups to lysine residues in histone tail domains, increasing the basicity of the lysine. Methylation of histone lysine residues in eukaryotes is most commonly catalyzed by methyltransferases in the SET domain superfamily (Fig. 1C), though it can also be catalyzed by the DOT1 family which lack SET domains (Khorasanizadeh, 2004). Lysine methylation predominantly occurs on histone H3 tails, and methylation of H3K4, H3K79, and H3K36 is associated with gene activation, while methylation of H3K9, H3K27, and H4K20 is associated with repressed gene expression (Sautel et al., 2007). Demethylation is carried out using Jumonji-related domain containing proteins (JmjC) (Iyer et al., 2008). Cryptosporidium parvum encodes several SET domain-containing proteins (Table 2) but does not encode any JmjC domain containing proteins (Sawant et al., 2022). Gastric species of Cryptosporidium including C. andersoni and C. muris encode a DOT1 family methyltransferase that is absent in C. parvum (Sawant et al., 2022).Table 2SET domain-containing proteins in Cryptosporidium parvum.Table 2. Gene IDProtein nameaPredicted targetaLife cycle stagebSingle cell atlas designationbcpbgf_8002730/cgd8_2730SET1H3K4AsexualClusters 3, 4, 10, 13cpbgf_500400/cgd5_400SET2H3K36Not stage-specificClusters 2, 3, 10, 13cpbgf_400370/cgd4_370SET8H4K20Not stage-specific–cpbgf_4002090/cgd4_2090AKMTPossible non-histone methyltransferaseSexualClusters 7, 8cpbgf_5002340/cgd5_2340KMToxH4/H2ANot stage-specific–cpbgf_7005090/cgd7_5090SET Unk1H3K4Asexual–cpbgf_1002170/cgd1_2170SET Unk2H3K4Asexual–cpbgf_6001470/cgd6_1470SET Unk3H3K27AsexualCluster 2aThe SET domain family methyltransferases in C. parvum were originally characterized by Sawant et al. (2022).bSingle-cell transcriptome atlas designation and life cycle stage where each protein was highly expressed was originally published by Walzer et al. (2024). Clusters 1–9 correspond to asexual development, 10–12 correspond to male sexual development, and 13–18 correspond to female sexual development.
Lactylation
2.1.3
Lactylation of histones was recently detected in Plasmodium falciparum as an additional mechanism of post-translational histone modification potentially involved in epigenetic regulation (Merrick, 2023). In lactylation, a lactyl group is added to lysine residues in histones. Lactylation of histones is generally considered to be a mark of activation and was only recently discovered in humans (Zhang et al., 2019). In humans, and in P. falciparum, lactylation levels fluctuate with lactate levels, providing potential environmental cues. In P. falciparum, hyperlactatemia is a strong predictor of severe malarial disease (Possemiers et al., 2021). Cryptosporidium parvum does contain a bacterial-type cytosolic and parasitophorous vacuole membrane associated L-lactate dehydrogenase (LDH) that can produce lactate (Zhang et al., 2015). Currently, it is unknown if lactylation of histones exists in C. parvum and thus may play a role in epigenetic regulation.
Chromatin remodelling
2.2
Enzymes involved in chromatin remodelling typically utilize NTP hydrolysis and usually contain P-loop NTPase folds (Iyer et al., 2008). There are two main classes of these enzymes: SWI2/SNF2 ATPases which perform local remodelling by altering nucleosome positioning, and the SMC ATPases which belong to the ABC superfamily (Iyer et al., 2008). The C. parvum reference genome has several annotated SWI2/SNF2 and SMC ATPases that possibly have roles in chromatin remodelling but have not been the subject of functional investigation. Some Myb domain containing proteins may have roles in chromatin remodelling (see Section 3.3); however, these predictions are based on homology to proteins in other organisms and lack functional confirmation in Cryptosporidium spp.
Cytosine methylation
2.3
In higher eukaryotes, CpG dinucleotides are often a target for covalent attachment of methyl groups, where the methyl group protrudes from the cytosine nucleotide into the major groove. The effect is two-pronged; transcription factors are displaced, and the attraction of methyl-binding proteins is associated with gene silencing and chromatin compaction (Fazzari and Greally, 2004). Cytosine-5 DNA methyltransferases catalyze the attachment of methyl groups to cytosine, and this family of enzymes is conserved in most eukaryotes. Cryptosporidium parvum encodes one annotated cytosine-5 DNA methyltransferase, DNMT2 (cpbgf_5002100/cgd5_2100), which is most likely involved in RNA modifications (see Section 4.2). In some eukaryotes, cytosine methylation is not always localized to CpG dinucleotides (Fisher et al., 2004), though genome-wide assessments using several detection methods have demonstrated that C. parvum has no detectable cytosine methylation (Gissot et al., 2008). While this trait was initially thought to be shared with Toxoplasma gondii and P. falciparum (Choi et al., 2006; Gissot et al., 2008), more recent investigations demonstrated low levels of cytosine methylation in P. falciparum (Gissot et al., 2008; Lucky et al., 2023) and T. gondii (Wei et al., 2017). Cytosine methylation in DNA is not considered a major contributor to epigenetic processes in Cryptosporidium spp.
Transcription factors
3
ApiAP2 family
3.1
The apicomplexan AP2 (ApiAP2) transcription factor family members are the most extensively studied gene regulatory components across the Apicomplexa (Fig. 2A). Structurally, the AP2 protein domains that define the ApiAP2 transcription factors are ∼60 amino acids in length, with three highly conserved β-strands followed by a less strongly conserved α-helix (Balaji et al., 2005; De Silva et al., 2008). There are 18 proteins encoded by the C. parvum genome that are predicted to contain at least one AP2 domain (Oberstaller et al., 2014). ApiAP2 protein domains in C. parvum have been experimentally confirmed to bind a range of DNA motifs summarized in Table 3. While the ApiAP2 family of transcription factors resemble the Apetala 2/Ethylene Responsive Factor (AP2/ERF) group of transcription factors found in plants, there are many additional family members in apicomplexans. There is also greater sequence diversity in the ApiAP2 transcription factor family, and consequently a wider repertoire of DNA motifs that they will bind (Balaji et al., 2005). Phylogenetic analyses of AP2 domain sequences across the SAR clade (Burki et al., 2020), encompassing the Stramenopila, Alveolata and Rhizaria lineages (Grattepanche et al., 2018), have indicated that the AP2 domains in apicomplexans are more closely related to the AP2 domains in Perkinsozoa than the Dinoflagellata (Oberstaller et al., 2014). There are four AP2 domains across three C. parvum proteins that likely existed in the myzozoan common ancestor, though lost in the dinoflagellates (Oberstaller et al., 2014). Ten AP2 domains across nine C. parvum proteins have homologs in most apicomplexans but are absent outside of the phylum, and ten AP2 domains across eight C. parvum proteins are specific to C. parvum (Oberstaller et al., 2014), indicating that most of their ApiAP2 transcription factors are the result of lineage-specific expansions, which is also the case for the genera Plasmodium and Toxoplasma (Balaji et al., 2005). Within the genus Cryptosporidium, most AP2 domains are conserved, but there is variation between species. For example, C. muris and C. andersoni are each missing five domains and additionally have two differences between them. Domain confirmation in all species will require gapless genome assemblies.Fig. 2. Possible transcription factors in C. parvum that may have a role in gene regulation. Little is known about the entire transcription factor complexes that regulate gene expression in Cryptosporidium spp. Schematic diagrams representing our understanding of ApiAP2 transcription factor activation of gene expression in Cryptosporidium spp. (A), E2F/DP transcription factor activity in eukaryotes (B), possible Myb/SANT gene regulation in Cryptosporidium spp. (C), and possible role of C2H2 ZnF in Cryptosporidium spp. gene regulation (D). Only one transcription factor protein for each scenario is shown but several may be present. Created in BioRender. Gunasekera, S. (2025) https://BioRender.com/odz641n.Fig. 2. Table 3List of overrepresented putative DNA-binding motifs in Cryptosporidium parvum upstream promoter regions.Table 3. Motif familyMotif sequenceReferenceTrans factorAP2_15′-TGCATGCA-3′Bankier et al. (2003); Oberstaller et al. (2013)ApiAP2 (Table 4)AP2_25′-GCACAC-3′Oberstaller et al. (2013)ApiAP2 (Table 4)G-box5′-G.GGGG-3′Mullapudi et al. (2007); Cohn et al. (2010); Oberstaller et al. (2013)ApiAP2 (Table 4)E2F5′-[A/T][C/G]GCGC[G/C][A/T]-3′Bankier et al. (2003); Oberstaller et al. (2013)E2F/DP (Table 5)GAGA5′-GAGAGAGA-3′Oberstaller et al. (2013)UnknownCAAT-box5′-GGCCAATCT-3′Oberstaller et al. (2013)bZIP (not reviewed here)
Expression of many members of the ApiAP2 transcription factor family in C. parvum appears to be stage-specific (Table 4), and there is considerable evidence indicating that they may play a large role in male and female fate determination (Tandel et al., 2019, 2023; Hasan et al., 2024; Walzer et al., 2024). The regulatory targets of most of the ApiAP2 transcription factors remain unknown, except for AP2-F, which has been demonstrated to regulate the expression of six proteins of the crystalloid body (cgd8_4290, cgd7_5140, cgd7_300, cgd7_1730, cgd2_790, cgd2_2110) and cgd7_5050 (NIMA kinase) (Tandel et al., 2023).Table 4. List of ApiAP2 domain-containing proteins in *Cryptosporidium parvum.*Table 4. Gene IDaLife cycle stageSingle-cell atlas designationbPrimary binding motifEvolutionary cladeacpbgf_8003230/cgd8_3230Asexual (Tandel et al., 2019; Walzer et al., 2024)Cluster 1AP2_1-like (Oberstaller et al., 2013, 2014)Ancestralcpbgf_400600/cgd4_600Not stage-specific (Tandel et al., 2019; Walzer et al., 2024)Cluster 6AP2_2-like (Oberstaller et al., 2013, 2014)Pan-apicomplexancpbgf_5002570/cgd5_2570Asexual (Walzer et al., 2024), in vivo female (Tandel et al., 2019)Clusters 6, 75′-GTGTGT-3′ (Oberstaller et al., 2014)Pan-apicomplexancpbgf_8003130/cgd8_3130Asexual (Mauzy et al., 2012; Walzer et al., 2024), in vivo female (Tandel et al., 2019)Cluster 7AP2_2-like (Oberstaller et al., 2013, 2014)Ancestralcpbgf_1003520/cgd1_3520Not stage-specific (Walzer et al., 2024)Cluster 7AP2_1-like (Oberstaller et al., 2013, 2014)Pan-apicomplexancpbgf_4002950/cgd4_2950Asexual (Mauzy et al., 2012; Walzer et al., 2024), in vivo female (Tandel et al., 2019)Cluster 85′-GCGTGCA-3′ (Oberstaller et al., 2014)Cryptosporidium-specificcpbgf_3002970/cgd3_2970Male-specific (Walzer et al., 2024)Clusters 10, 11Does not bind DNA (Oberstaller et al., 2014)Cryptosporidium-specificcpbgf_6002670/cgd6_2670 (AP2-M)Male-specific (Tandel et al., 2023; Walzer et al., 2024)Cluster 115′-AAAA-3′ (Oberstaller et al., 2014)Cryptosporidium-specificcpbgf_2003490/cgd2_3490Female-specific (Mauzy et al., 2012; Tandel et al., 2019; Hasan et al., 2024; Walzer et al., 2024)Clusters 15-18AP2_1-like (Oberstaller et al., 2013, 2014)Pan-apicomplexancpbgf_4001110/cgd4_1110 (AP2-F)Female-specific (Mauzy et al., 2012; Tandel et al., 2019, 2023; Walzer et al., 2024)Clusters 16, 17AP2_1-like (Oberstaller et al., 2013, 2014)Ancestralcpbgf_800810/cgd8_810Female-specific (Tandel et al., 2019; Walzer et al., 2024)Clusters 16-18G-box (Oberstaller et al., 2013, 2014)Pan-apicomplexancpbgf_6005320/cgd6_5320Not stage-specific (Tandel et al., 2019; Walzer et al., 2024)–Does not bind DNA (Oberstaller et al., 2014)Pan-apicomplexancpbgf_3001980/cgd3_1980Not stage-specific (Tandel et al., 2019; Walzer et al., 2024)–Does not bind DNA (Oberstaller et al., 2014)Pan-apicomplexancpbgf_5004250/cgd5_4250Not stage-specific (Tandel et al., 2019; Walzer et al., 2024)–AP2_1-like (Oberstaller et al., 2013, 2014)Pan-apicomplexancpbgf_6001140/cgd6_1140Not stage-specific (Walzer et al., 2024)–Does not bind DNA (Oberstaller et al., 2014)Cryptosporidium-specificcpbgf_4003820/cgd4_3820Not stage-specific (Walzer et al., 2024)–AP2_1-like (Oberstaller et al., 2013, 2014)Pan-apicomplexancpbgf_6002600/cgd6_2600Not stage-specific Tandel et al. (2019)–5′-GTGTGT-3′ (Oberstaller et al., 2014)Cryptosporidium-specificcpbgf_2002990/cgd2_2990––G-box (Oberstaller et al., 2014)Cryptosporidium-specificaIdentification of ApiAP2 domain-containing proteins and kingdom-wide evolutionary analysis of AP2 domains was originally published by Oberstaller et al. (2014).bSingle-cell transcriptome atlas designation was originally published by Walzer et al. (2024). Clusters 1–9 correspond to asexual development, 10–12 correspond to male sexual development, and 13–18 correspond to female sexual development.
E2F family
3.2
Cryptosporidium thus far, appears to be the only genus in the Apicomplexa to have retained the E2F/DP transcription factor family (Templeton et al., 2004; Oberstaller et al., 2013; Baptista et al., 2025). The E2F transcription factor family comprises two subfamilies: E2F and DP. In higher eukaryotes, one member of each of these subfamilies come together to form an active heterodimer that binds promoters to regulate the expression of many target genes, where they can act as either transcription activators or repressors (Fig. 2B). E2F transcription activity in higher eukaryotes can be modulated through complex formation with other regulatory proteins (Attwooll et al., 2004). The C. parvum genome encodes two E2F transcription factors and two DP binding partners (Table 5), which peak in expression at 2 h and 12 h post-infection (Mauzy et al., 2012). It remains unknown whether C. parvum encodes any other proteins that interact with the E2F transcription factors, or how they may utilize E2F transcription factors for gene regulation. Of interest, E2F motifs are the most abundant transcription factor binding site in C. parvum (Oberstaller et al., 2013) and have been recently suggested to be an important regulator of early asexual development and polycistronic transcripts (Xiao et al., 2025). E2F motifs are overrepresented upstream of genes associated with DNA replication and glycolysis (Oberstaller et al., 2013), and in the internal transcribed spacer of genes present in polycistronic transcripts (Xiao et al., 2025). E2F motifs are also overrepresented upstream of co-regulated ribosomal protein encoding genes in C. parvum, starkly differing from the G-box motif found in other Apicomplexa (Oberstaller et al., 2013), lending further support to extensive differences in gene regulation mechanisms in the genus Cryptosporidium.Table 5. List of E2F/DP transcription factors in *Cryptosporidium parvum.*Table 5. Gene IDProtein subfamilyLife cycle stageSingle-cell atlas designationaBinding motifbcpbgf_1001560/cgd1_1560E2FAsexualClusters 1, 25′-[A/T][C/G]GCGC[G/C][A/T]-3′cpbgf_6001430/cgd6_1430E2FAsexualClusters 1, 25′-[A/T][C/G]GCGC[G/C][A/T]-3′cpbgf_7003650/cgd7_3650DPAsexualClusters 1, 25′-[A/T][C/G]GCGC[G/C][A/T]-3′cpbgf_8001850/cgd8_1850DPAsexualClusters 1, 25′-[A/T][C/G]GCGC[G/C][A/T]-3′aSingle-cell transcriptome atlas designation was originally published by Walzer et al. (2024), and mined for E2F/DP expression by Xiao et al. (2025).bThe E2F binding motif in C. parvum was originally reported by Bankier et al. (2003) and investigated further by Oberstaller et al. (2013).
Myb transcription factors
3.3
The Myb transcription factor superfamily is defined by the Myb DNA binding domain that contains between one to four Myb repeats stretching 50–53 amino acids in length each, termed R1, R2, R3, and R4. Each Myb repeat contains three alpha helices, the second and third of which confers the helix-turn-helix secondary structure that enables DNA binding. The Myb superfamily is subdivided into four groups (1R-Myb, 2R-Myb, 3R-Myb, and 4R-Myb) based on the number of Myb repeats it possesses and their position within the Myb domain. Importantly, a subgroup of 1R- and 2R Mybs termed the SANT domain proteins cannot bind DNA and instead interact with histone tails (Fig. 2C), often as part of multimeric protein complexes (Prouse and Campbell, 2012). The C. parvum genome has 16 annotated Myb domain-containing proteins (Table 6). The diverse roles of Mybs in apicomplexans have been recently reviewed comprehensively (Schwarz and Lourido, 2023). The only Myb in C. parvum that has been functionally validated is Myb-M (cpbgf_6002250/cgd6_2250), which is the earliest transcription factor that controls male sexual fate determination in C. parvum (Walzer et al., 2024).Table 6. Myb domain-containing proteins in Cryptosporidium parvum.Table 6. Gene IDaSuggested role in gene regulationaMyb subfamilyaLife cycle stagebSingle cell atlas designationbcpbgf_6004510/cgd6_4510DNA binding1R-MybNot stage-specificClusters 3, 4, 10cpbgf_3002510/cgd3_2510DNA binding1R-MybNot stage-specific–cpbgf_5001120/cgd5_1120Histone modification1R-SANTNot stage-specific–cpbgf_4001270/cgd4_1270Histone modification1R-SANTNot stage-specific–cpbgf_8004840/cgd8_4840––AsexualCluster 1cpbgf_1002330/cgd1_2330Histone modification1R-SANTNot stage-specificClusters 4–6, 10–12cpbgf_8002770/cgd8_2770Chromatin remodellingISW1Female-specificCluster 18cpbgf_6003860/cgd6_3860Chromatin remodellingISW1Male-specificCluster 10cpbgf_500110/cgd5_110DNA bindingCDC5LNot stage-specificClusters 3, 7, 11, 13cpbgf_6002250/cgd6_2250 (Myb-M)Male sexual fate determination1-3R-MybMale-specificClusters 10, 11cpbgf_2003980/cgd2_3980Histone modification1R-SANTNot stage-specificClusters 8, 14cpbgf_2002260/cgd2_2260Displacement of polycomb-repressive complexDNAJCAsexualClusters 1, 9cpbgf_2003460/cgd2_3460–CHYNot stage-specific–cpbgf_2001740/cgd2_1740Chromatin remodellingSWI3Not stage-specificClusters 3, 4, 10, 13, 14cpbgf_400880/cgd4_880Histone acetyltransferaseAda2Not stage-specificClusters 9, 12cpbgf_3001120/cgd3_1120DNA binding4R-MybNot stage-specificClusters 9, 11aMyb domain-containing proteins in the Apicomplexa have been reviewed in detail by Schwarz and Lourido (2023). Note that suggested roles in gene regulation are based on sequence homology only and have not been functionally investigated.bSingle-cell transcriptome atlas designation and the highest expressed life cycle stage was originally published by Walzer et al. (2024). Clusters 1–9 correspond to asexual development, 10–12 correspond to male sexual development, and 13–18 correspond to female sexual development.
C2H2 zinc finger proteins
3.4
Zinc finger domains (ZnF) are among the most widespread DNA binding domains in eukaryotes. They bind zinc ions most commonly via pairs of cysteine and histidine residues (C2H2 ZnF). C2H2 ZnF have a beta-beta-alpha secondary structure, and the basic and hydrophobic residues in the alpha helix confer DNA binding capability. A single C2H2 ZnF cannot typically function alone to regulate transcription, and typically a series of three or more C2H2 ZnF will bind cis regulatory elements (Matthews and Sunde, 2002). While C. parvum encodes several putative C2H2 ZnFs (Table 7), their DNA binding sites and their role in gene regulation in Cryptosporidium spp. remains uncharacterized (Fig. 2D).Table 7. Annotated C2H2 ZnF domain-containing proteins in Cryptosporidium parvum.Table 7. Gene IDaLife cycle stagebSingle cell atlas designationbcpbgf_2001150/cgd2_1150Not stage-specificClusters 2, 3, 10, 13cpbgf_3001060/cgd3_1060Not stage-specific–cpbgf_3001440/cgd3_1440Not stage-specific–cpbgf_5001110/cgd5_1110Not stage-specific–cpbgf_7001170/cgd7_1170Not stage-specific–cpbgf_7004300/cgd7_4300AsexualClusters 8, 9cpbgf_7005380/cgd7_5380Not stage-specificClusters 3–5, 10, 13–15cpbgf_8001550/cgd8_1550AsexualClusters 1, 6aC2H2 ZnF domain-containing proteins were mined from the updated C. parvum genome annotation in Baptista et al. (2025).bSingle-cell transcriptome atlas designation and the highest expressed life cycle stage was originally published by Walzer et al. (2024). Clusters 1–9 correspond to asexual development, 10–12 correspond to male sexual development, and 13–18 correspond to female sexual development.
Non-coding RNAs
4
There are several types of non-coding RNAs (ncRNAs) and many play roles in gene expression at the level of epigenetics, transcription, transcript processing and translation. The role of ncRNAs in apicomplexans has been reviewed in detail elsewhere (Li et al., 2020; Mitesser et al., 2024). In C. parvum, ncRNAs are abundant (Li et al., 2021, 2022), and approximately 10% of the genes in C. parvum have an associated antisense transcript of unknown function (Li et al., 2021; Baptista et al., 2025).
Long non-coding RNAs
4.1
Long non-coding RNAs (lncRNAs) are defined as transcripts longer than 200 nucleotides with open reading frames shorter than 30 amino acids that do not encode a known protein product. Several ncRNAs, especially lncRNAs, have been well characterized in the apicomplexan parasite genera Plasmodium and Toxoplasma, where they have been implicated in epigenetic regulation of numerous genes and processes, including var gene expression (Jing et al., 2018), telomere-associated repetitive elements (TARE) (Broadbent et al., 2011), and translational blocking (Eksi et al., 2012). In C. parvum, numerous lncRNAs have been annotated and studied for patterns of developmentally regulated gene expression (Li et al., 2021; Baptista et al., 2025). In the C. parvum IOWA II BGF Telomere-to-Telomere genome assembly (Baptista et al., 2025), 766 lncRNAs have been annotated based on gene expression data. Natural antisense transcripts are a subtype of lncRNA that either partially or entirely, overlap a corresponding sense transcript. They have been reported extensively in P. falciparum and T. gondii (Radke et al., 2005; Siegel et al., 2014), though their function is not well-understood. Existing evidence indicates that most annotated lncRNAs in C. parvum fall under the definition of a natural antisense transcript (Li et al., 2021). Given that C. parvum lacks genes that encode Dicer and Argonaute (Abrahamsen et al., 2004), natural antisense transcripts likely exert their influence on gene regulation in ways other than triggering dsRNA degradation (Abrahamsen et al., 2004). The only functionally characterized lncRNAs in C. parvum are those that are exported and influence host gene expression (Wang et al., 2016, 2017; Ming et al., 2017).
Small ncRNAs and epitranscriptomics
4.2
Very little is known about RNA modifications in the genus Cryptosporidium, especially those that may affect gene expression (Fig. 3). We have non-validated evidence of RNA modifications in C. parvum based on Oxford Nanopore Direct RNAseq data, but the exact chemical modifications are computational predictions at this time (unpublished data). One gene encoding an ortholog of the methyltransferase DNMT2 protein (cpbgf_5002100/cdg5_2100), is annotated in the C. parvum reference genome (Abrahamsen et al., 2004; Baptista et al., 2025). DNMT2 is involved in tRNA methylation and possibly other RNA modifications (Lucky et al., 2023) and has been studied in other apicomplexans but not in Cryptosporidium spp.Fig. 3. Possible epitranscriptomic modifications that Cryptosporidium spp. could utilize for gene regulation. Small RNAs are shown in blue, methyl groups are shown in red, modified regions of target RNAs are shown in green. Created in BioRender. Gunasekera, S. (2025) https://BioRender.com/9bqfw87.Fig. 3
Pseudouridinylation is a common post-transcriptional modification of RNA molecules in eukaryotes that results in the isomerization of uridine to pseudouridine. Pseudouridinylation can be catalyzed by ribonucleoproteins (RNPs) containing small nucleolar RNAs (snoRNAs) with H/ACA boxes that direct the position of pseudouridinylation in the RNA molecule (Charpentier et al., 2005). Pseudouridinylation in T. gondii has been functionally investigated and found to be developmentally regulated and to have a small but statistically significant effect on mRNA stability (Nakamoto et al., 2017). Cryptosporidium parvum contains orthologs of many genes encoding proteins in the H/ACA RNP complex as well as four H/ACA box snoRNAs (Table 8). The 2′-O-methylation of rRNA molecules can also be catalyzed by RNPs containing snoRNAs, with the exception that the snoRNAs contain C/D boxes (SNORDs) that direct the position of the post-transcriptional modification (Charpentier et al., 2005). Cryptosporidium parvum contains 5 identified C/D box snoRNAs (Table 8). The biological significance and modification targets in Cryptosporidium spp. are not experimentally confirmed for either class of modifications.Table 8. List of H/ACA RBPs and SNORDs in Cryptosporidium parvum.Table 8. Gene IDModification/functioncpbgf_100530/cgd1_530H/ACA ribonucleoprotein complex subunit Nop10cpbgf_5001760/cgd5_1760H/ACA ribonucleoprotein complex subunitcpbgf_8001060/cgd8_1060H/ACA ribonucleoprotein complex subunit Gar1/Naf1cpbgf_5001585/cgd5_1585H/ACA snoRNAcpbgf_7002033/cgd7_2033H/ACA snoRNAcpbgf_7003425/cgd7_3425H/ACA snoRNAcpbgf_7005104/cgd7_5104H/ACA snoRNAcpbgf_2003583/cgd2_3583C/D snoRNAcpbgf_5003935/cgd5_3935C/D SNORD96cpbgf_6003219/cgd6_3219C/D snoRNAcpbgf_7001545/cgd7_1545C/D snoRNAcpbgf_7005107/cgd7_5107C/D SNORD36
Upstream open reading frames
5
Translation in eukaryotes is initiated by the translation pre-initiation complex, which is recruited to the 5′ cap of the mRNA molecule before scanning downstream for an AUG start codon. Recognition of the start codon is heavily influenced by the surrounding Kozak sequence. In some organisms, the translation pre-initiation complex can also be recruited independently of the 5′ cap at internal ribosomal entry sites (IRES) (Karginov et al., 2017). Furthermore, translation initiation can be affected by the presence of an upstream open reading frame (uORF) where the 5′ untranslated region of an mRNA molecule also contains a start codon, with an in-frame stop codon that can be either upstream or within the main CDS (Fig. 4A). The presence of an uORF usually represses translation of the main CDS by either reducing efficiency of translation initiation at the main AUG or by triggering mRNA decay. Leaky ribosomal scanning can also occur, where the first translation initiation site may have a weaker Kozak sequence, resulting in the translation pre-initiation complex bypassing the first AUG (Dueñas et al., 2025). The presence of uORFs has been detected at an unusually high frequency in the genera Toxoplasma and Plasmodium, where almost all Toxoplasma and Plasmodium transcripts with an annotated 5′ UTR contain a uORF, reviewed in detail elsewhere (Kaur and Patankar, 2021). The presence of uORFs has not been described in Cryptosporidium spp., though future investigations are aided by the recent C. parvum IOWA-BGF Telomere-to-Telomere assembly with updated UTR annotations (Baptista et al., 2025).Fig. 4. The structure of uORFs and polycistronic transcripts. Schematic diagram of a transcript with an uORF (not yet reported in Cryptosporidium spp.) (A), and a polycistronic transcript, recently reported in C. parvum (Xiao et al., 2025) (B). Created in BioRender. Gunasekera, S. (2025) https://BioRender.com/Uizemxv.Fig. 4
The recent discovery of polycistronic transcripts in C. parvum sporozoites may represent a potentially related gene regulatory phenomenon where, instead of a short uORF being present in the 5′ UTR, an entire protein-encoding gene of unrelated function is transcribed upstream of a second ORF (Fig. 4B). The polycistronic transcripts most commonly consist of two genes and are termed dicistrons, but tri- and quad-cistronic genes are detected and were confirmed with RT-PCR. To date, 201 polycistronic transcripts have been detected in two different strains of C. parvum, in both sporozoites and merozoites (Xiao et al., 2025). Approximately 400 genes, 10% of the protein-encoding repertoire, are observed in polycistronic transcripts. The role of polycistronic transcription in gene regulation at either the transcriptional or post-transcriptional level is yet to be explored in Cryptosporidium spp. In other eukaryotes, polycistrons have been associated with translational control via IRES (Karginov et al., 2017) or leaky ribosomal scanning (Dueñas et al., 2025).
R-loops
6
R-loops are DNA:RNA hybrids that can accumulate at specific regions in the genome. They have been associated with gene regulation via class switching in immunoglobulins, inhibition of transcription, protection from methylation and open chromatin (Roy et al., 2008; Ginno et al., 2012; Castellano-Pozo et al., 2013; Costantino and Koshland, 2015; D'Souza et al., 2018). R-loops have not yet been experimentally confirmed in Cryptosporidium spp., but the abundance of lncRNAs raises the possibility. R-loops have been detected in P. falciparum in association with TAREs, reviewed in (Simantov et al., 2022).
Concluding remarks
7
The genome sequences for Cryptosporidium spp. encode genes for many of the key regulatory proteins and classes of RNAs that have been characterized in other apicomplexans. Yet, given the challenges of working with Cryptosporidium spp., the functions of these proteins and RNAs have not been experimentally validated. Cryptosporidium spp. also differ from other apicomplexans in that they have retained E2F transcription factors and C. parvum has evolved polycistronic transcription. The transcriptional and post-transcriptional regulatory networks that underlie the tight coordination of gene expression in the genus Cryptosporidium remain largely unexplored. Recent genetic and technological advancements in Cryptosporidium spp. are aiding the community's ability to study gene regulation (Tandel et al., 2023), hopefully moving many of the unknowns into knowns, rapidly. The explosion of new data and insights will facilitate our understanding of gene regulation and biology in Cryptosporidium spp. further.
CRediT authorship contribution statement
Samantha Gunasekera: Writing – original draft, Writing – review & editing, Visualization. Jessica C. Kissinger: Conceptualization, Writing – original draft, Writing – review & editing.
Ethical approval
Not applicable.
Data availability
Not applicable.
Funding
This work was supported in part by grants from the National Institutes of Health, National Institute of Allergy and Infectious Diseases: R21AI80871 and NIH R01AI148667.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abrahamsen M.S.Templeton T.J.Enomoto S.Abrahante J.E.Zhu G.Lancto C.A.Complete genome sequence of the apicomplexan, Cryptosporidium parvum Science 30420044414451504475110.1126/science.1094786 · doi ↗ · pubmed ↗
- 2Ammar R.Torti D.Tsui K.Gebbia M.Durbic T.Bader G.D.Chromatin is an ancient innovation conserved between Archaea and Eukaryae Life 12012 e 0007810.7554/e Life.00078 PMC 351045323240084 · doi ↗ · pubmed ↗
- 3Attwooll C.Denchi E.L.Helin K.The E 2F family: Specific functions and overlapping interests EMBO J.232004470947161553838010.1038/sj.emboj.7600481 PMC 535093 · doi ↗ · pubmed ↗
- 4Balaji S.Babu M.M.Iyer L.M.Aravind L.Discovery of the principal specific transcription factors of Apicomplexa and their implication for the evolution of the AP 2-integrase DNA binding domains Nucleic Acids Res.332005399440061604059710.1093/nar/gki 709PMC 1178005 · doi ↗ · pubmed ↗
- 5Bankier A.T.Spriggs H.F.Fartmann B.Konfortov B.A.Madera M.Vogel C.Integrated mapping, chromosomal sequencing and sequence analysis of Cryptosporidium parvum Genome Res.132003178717991286958010.1101/gr.1555203 PMC 403770 · doi ↗ · pubmed ↗
- 6Baptista R.P.Li Y.Sateriale A.Sanders M.J.Brooks K.L.Tracey A.Ansell B.R.E.Long-read assembly and comparative evidence-based reanalysis of Cryptosporidium genome sequences reveal expanded transporter repertoire and duplication of entire chromosome ends including subtelomeric regions Genome Res.3220222032133476414910.1101/gr.275325.121PMC 8744675 · doi ↗ · pubmed ↗
- 7Baptista R.P.Xiao R.Li Y.Glenn T.C.Kissinger J.C.New T 2T assembly of Cryptosporidium parvum IOWA II annotated with Legacy-Compatible Gene identifiers Sci. Data 12202510394053746410.1038/s 41597-025-05364-3PMC 12179316 · doi ↗ · pubmed ↗
- 8Bozdech Z.Llinas M.Pulliam B.L.Wong E.D.Jingchun Z.De Risi J.L.The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum P Lo S Biol.120038510010.1371/journal.pbio.0000005 PMC 17654512929205 · doi ↗ · pubmed ↗