Ropeginterferon alfa-2b as a promising alternative to conventional interferon in CDA type 1: a case report of two siblings
Tyng-Wei Yang, Xavier Cheng-Hong Tsai, Yu-Hsuan Fu, Liang-In Lin, Hsin-An Hou

TL;DR
A new long-acting interferon improved treatment for a rare anemia in two siblings, reducing the need for blood transfusions and side effects.
Contribution
First documented use of ropeginterferon alfa-2b in CDA-I patients, showing improved efficacy and tolerability.
Findings
Two CDA-I patients achieved transfusion independence after switching to ropeginterferon alfa-2b.
Biweekly dosing reduced injection burden and adverse effects compared to conventional interferon.
Hemoglobin levels improved while maintaining treatment efficacy.
Abstract
Congenital dyserythropoietic anemia type I (CDA-I) is a rare hereditary anemia caused by CDAN1 or C15orf41 mutations, with CDAN1-related cases responding to interferon-alpha (IFN-α) therapy. However, traditional IFN-α requires frequent injections and often causes flu-like symptoms, which can hinder long-term adherence. Here, we present the first documented use of ropeginterferon alfa-2b, a next-generation pegylated interferon, in two patients with CDA-I. Both patients, who were previously transfusion dependent, achieved transfusion independence and improved hemoglobin levels after transitioning from recombinant IFN-α to ropeginterferon alfa-2b with biweekly dosing. This treatment preserved efficacy while minimizing adverse effects and the injection burden. Our findings suggest that ropeginterferon alfa-2b may serve as a more tolerable and effective long-term treatment. Further…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
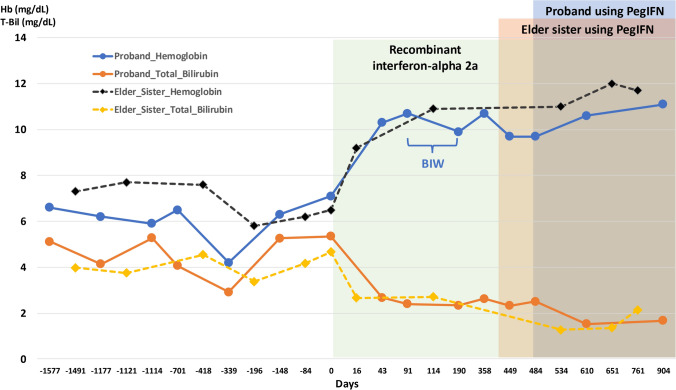 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEosinophilic Disorders and Syndromes · Immunodeficiency and Autoimmune Disorders · Neutrophil, Myeloperoxidase and Oxidative Mechanisms
Introduction
Congenital dyserythropoietic anemia (CDA) is a diverse group of inherited disorders characterized by ineffective erythropoiesis. CDA is classified into five types (CDA-I, II, III, transcription factor-related CDAs, and CDA variants) [1], which were historically distinguished based on clinical presentation and erythroblast morphological changes. In CDA-I, for example, erythroblasts typically exhibit a “spongy heterochromatin” or “Swiss cheese” appearance when examined by scanning electron microscopy (SEM). Advancements in molecular genetics have led to the identification of pathogenic genes responsible for CDA. CDA-I is caused by biallelic mutations in either the CDAN1 or C15orf41 gene. To date, 56 causative mutations have been documented, with 51 occurring in the CDAN1 gene and 5 in the C15orf41 gene [2].
CDA treatment was limited to supportive care for many years until 1995, when Lavabre-Bertrand et al. unexpectedly reported that interferon-alpha (IFN-α) was effective in treating CDA-I in a 28-year-old patient with concomitant CDA-I and hepatitis C [3]. However, its requirement for thrice-weekly subcutaneous injections can significantly impact quality of life. Here, we describe two CDA-I patients who achieved transfusion independence and improved hemoglobin levels with ropeginterferon alfa-2b, a next-generation pegylated interferon requiring less frequent administration. This treatment may offer a practical and well-tolerated option for managing CDA-I.
Case presentation
A 29-year-old woman presented to a hematology clinic with intermittent shortness of breath persisting for several years. Although she was diagnosed with alpha-thalassemia minor at birth, she remained asymptomatic throughout childhood. She had no skeletal or limb abnormalities, chest deformities, or short stature. Her first visit to a pediatric hematologist was at the age of 15, when microcytic anemia (hemoglobin 7.0 g/dL, MCV 71.7 fL) and indirect hyperbilirubinemia (total/direct bilirubin = 6.23/0.7 mg/dL) were documented (Table 1). At that time, genetic testing revealed a heterozygous alpha-globin gene deletion (-SEA), leading to a diagnosis of α-thalassemia. Her red blood cell morphology revealed only anisopoikilocytosis. Among her family members, including three siblings, only the proband and one elder sister exhibited significant hemolytic anemia, despite both the father and the same sister having α-thalassemia minor.Table 1. Laboratory values of the patientTestValueReference range15-year-oldBefore IFN-α treatment1 year after IFN-α treatmentRed blood cell count (M/µL)3.113.064.643.7–4.96Hemoglobin (g/dl)7.07.110.710.2–13.2Hematocrit (%)22.323.936.432.4–41.2Mean corpuscular volume (fL)71.778.178.476.9–93.7Platelet count (K/µL)237190163186–353Reticulocyte (%)1.071.350.9 ~ 1.94White blood cell count (K/µL)6.413.744.934.19–11.4Blast (%)000Promyelocyte (%)000Myelocyte (%)000Metamyelocyte (%)000Band neutrophil (%)0000–5Segmented neutrophil (%)61.162.954.141.6–74.4Eosinophil (%)1.92.41.40.3–7.9Basophil (%)0.20.50.20.2–1.6Monocyte (%)2.32.93.73.3–8.9Lymphocyte (%)34.531.340.63.3–8.9Normoblasts (/100WBC)000Total bilirubin (mg/dL)5.195.352.630.2–1.2Direct bilirubin (mg/dL)0.630.540.450–0.4Alanine aminotransferase (U/liter)71219 < 31Lactate dehydrogenase (U/liter)439248173230–460Hb-EP(HbH)(%)NegativeHb-HPLC(HbA2)(%)2.2 < 3.4Hb-HPLC(HbF)3.1 < 1.9
Since the age of 20, she began requiring regular blood transfusions every 2–4 months to alleviate exercise intolerance and dizziness and was prescribed folic acid supplementation. However, because her anemia was significantly more severe than expected for thalassemia minor alone, comprehensive testing, including hemoglobin electrophoresis, the eosin-5'-maleimide (EMA) binding test, the osmotic fragility test, and fluorescently labeled AERolysin (FLAER) flow cytometry, yielded normal results.
To further investigate the underlying cause of her hemolytic anemia, whole-exome sequencing followed by Sanger sequencing was performed. This analysis revealed compound heterozygous mutations in the CDAN1 gene: c.2140C > T (p.R714W), inherited from her mother, and c.2059C > T (p.R687C), inherited from her father. Her elder sister, who exhibited a similar clinical presentation, was also screened, with results revealing that she also carried the same heterozygous mutations. Based on these findings, the patient was diagnosed with α-thalassemia minor with CDA-I.
After confirming the diagnosis and having thorough discussions, we initiated treatment with recombinant interferon alfa-2a (Feronsure®) at a dose of 3 million IU three times per week. The patient demonstrated a significant response, with hemoglobin increasing from 7.1 g/dL to 10.3 g/dL and total bilirubin decreasing from 5.35 mg/dL to 2.68 mg/dL after six weeks of therapy (Fig. 1). However, frequent injections resulted in significant intolerance, including local injection site pain and fever, which adversely affected her quality of life. To mitigate these side effects, the Feronsure® dosage was tapered to two times per week, but this resulted in a decrease in hemoglobin levels. We sought an alternative to this long-term treatment which could last decades, as an approach to balance efficacy and tolerability. After 16 months on Feronsure®, we initiated treatment with ropeginterferon alfa-2b (Besremi®), which was administered biweekly, starting at a dose of 100 µg and gradually titrating up to 250 µg. The patient tolerated ropeginterferon alfa-2b well, with stable hemoglobin levels and no significant adverse effects. Notably, the same treatment approach was used for her older sister, who also showed a positive response and experienced a notable improvement in her quality of life.Fig. 1. Hemoglobin and total bilirubin (T-Bil) levels over time in the proband and her elder sister with CDA-I, with Day 0 marking the start of IFN treatment. The proband’s hemoglobin (blue solid line, circular markers) and total bilirubin (orange solid line, circular markers) improved after treatment initiation. The elder sister’s hemoglobin (black dashed line, diamond markers) and total bilirubin (yellow dashed line, diamond markers) levels followed similar trends. The green shaded area represents recombinant interferon-alpha 2a therapy, during which the proband’s dosing was adjusted to twice weekly owing to intolerance. The blue- and brown-shaded areas indicate the transition to ropeginterferon alfa-2b for the proband and her sister, respectively. Both patients demonstrated a sustained increase in hemoglobin and a decrease in T-Bil under ropeginterferon alfa-2b therapy (colour figure online)
Discussion
The diagnosis of rare inherited anemias, such as CDA-I, has traditionally depended on bone marrow examination and erythroblast morphology assessment, often leading to delayed recognition or misdiagnosis [4, 5]. Advances in next-generation sequencing have transformed this process, enabling earlier and more precise diagnoses while reducing reliance on invasive procedures. As molecular analysis becomes more accessible, genetic testing is now the preferred first-line tool for diagnosing CDA [1, 4], but biochemical and morphological assessments still play a role in resolving cases with uncertain genetic variants.
CDA is a group of rare congenital hypoproliferative anemias. Among its subtypes, CDA-I, an autosomal recessive disease caused by either biallelic CDAN1 or C15orf41 mutations, accounts for only 4.4% of all CDA cases [1]. Notably, CDA-I is the only CDA subtype that responds to IFN-α therapy. [6] Although the underlying mechanism remains unclear, a study by Wickramasinghe et al. revealed that CDA-I patients exhibit reduced IFN-α production compared with both healthy controls and patients with other CDA subtypes, possibly explaining its therapeutic benefit. [7] Table 2 summarizes previous CDA treatment approaches and their corresponding responses. Among CDA-I patients, the response to IFN-α varied, but the key observation is that continuous injections were necessary to maintain hemoglobin levels. Since the first report of IFN-α as an effective treatment for CDA-I in 1995, [3] its use has been limited by the need for thrice-weekly or twice-weekly injections, which affects long-term adherence. Subsequent reports have documented similar therapeutic benefits in CDA-I patients, further supporting its role as a disease-specific treatment [8–11]. While both CDAN1 and C15orf41 mutations cause CDA-I, a study reported that only patients with CDAN1 mutations respond well to IFN-α treatment, whereas those with C15orf41 mutations may not [12]. This difference could explain why not all CDA-I patients respond equally to IFN-α therapy. The differential interferon responsiveness between patients with CDAN1 and C15orf41 mutations remains incompletely understood. CDAN1 encodes Codanin-1, a protein implicated in chromatin assembly and nuclear envelope stability during erythropoiesis [13]. Dysregulation of Codanin-1 may render erythroid precursors more sensitive to the transcriptional and epigenetic modulation triggered by IFN-α. In contrast, C15orf41 is predicted to encode an endonuclease with potential roles in DNA repair or the DNA replication stress response [12]. These pathways may be less amenable to modulation by interferon signaling. Functional studies exploring interferon-induced transcriptional changes in CDAN1- versus C15orf41-mutated cells are needed to substantiate this hypothesis. However, because of the limited number of cases and the low availability of genetic testing, it remains unclear whether this difference in response rates is conclusive. While traditional IFN-α effectively treats CDA-I, its frequent dosing regimen poses significant quality-of-life challenges. Ropeginterferon Alfa-2b (Besremi®), a next-generation pegylated interferon, offers a longer half-life, allowing for biweekly administration.Table 2A brief review of prior treatment approaches for CDA and their corresponding responsesReferencesAgeSexCDA typeDx methodTx agentDoseHb (g/dL)AddendumBeforeAfterLavabre-Bertrand et al.[9] (1995)28FISEMIFN-alpha 2a3MU TIW × 24 wks8.611.5Responsive but requiring continuous treatmentWickramasinghe et al.[11]30FILMIFN-alpha 2a3 M IU TIW × 5 wks8.212.1ResponsiveShamseddine et al.[23]31MILMIFN-alpha3 M IU TIW × 16 wks8.412Responsive. The patient stopped the drug spontaneously. Hb remained stable for another 2 months then gradually dropped to 8.5 g/dL at 14 months after treatment initiation64FILMIFN-alpha3 M IU TIW × 12 wks8.811.0Responsive, but the Hb level dropped to pretreatment level after discontinuing IFN-alpha for 7 monthsMarwaha et al.[6]2.5FILMIFN-alpha 2b4.2 M IU/m^2^ TIW x 13wks8.54.0No response on Hb, transfusion requirements, or the size of the spleen and liver7MILMIFN-alpha 2b3.9 M IU/m^2^ TIW × 15 wks 6.5 M IU/m^2^ TIW × 15 wks8.09.011.5MILMIFN-alpha 2b2.6 M IU/m^2^ TIW × 9 wks 2.6 M IU/m^2^ QIW × 10 wks9.08.05.5MIILM, Ham’s testIFN-alpha 2b4.3 M IU/m^2^ TIW × 8 wks 4.3 M IU/m^2^ QIW × 4 wks4.04.06 mFIILM, Ham’s testIFN-alpha 2b5 M IU/m^2^ TIW × 6 wks 5 M IU/m^2^ QIW × 17 wks8.55.58MIILM, Ham’s testIFN-alpha 2b4.6 M IU/m^2^ TIW × 15 wks10.04.0Agrigento et al.[8]15FISEMIFN-alpha 2b4.5 M IU QW8.59.44Increased Hb level but hemolysis persisted This patient is also a Sicilian Beta Thalassemia carrier50FISEMIFN-alpha 2b9 M IU QW6.627.17Partial response with prolonged blood transfusion interval (30 days to 49 days) This patient is also a Sicilian Beta Thalassemia carrierRathe et al.[10]11 mMIC15orf41 (p.Leu178Gln)Peg-INF-alfa-2a45mcg QW × 36 wks9.810Stable hemoglobin and ferritin levelAbbreviations: IFN = Interferon; SEM = Scanning electron microscope; LM = Light microscope; TIW = Thrice weekly; QIW = Four times weekly; Ham’s test = Acidified serum lysis test
Pegylated interferon alfa-2a has demonstrated comparable or superior efficacy and a similar safety profile to conventional IFN-α in various disease settings, including chronic hepatitis C [14]. Ropeginterferon alfa-2b demonstrated greater drug exposure with a comparable safety profile to pegylated IFN alfa-2a in a phase I trial [15]. In patients with polycythemia vera, ropeginterferon also achieved a faster therapeutic response and was associated with significantly fewer flu-like side effects than peginterferon alfa-2a [16]. Given its enhanced efficacy and improved tolerability, ropeginterferon alfa-2b represents a preferred option when the immunomodulatory properties of IFN-α are desired.
In addition to its extended half-life and improved tolerability, ropeginterferon alfa-2b has pleiotropic immunomodulatory effects that may contribute to its therapeutic effect on CDA-I. Interferon-α is known to induce a wide array of interferon-stimulated genes involved in hematopoietic regulation, antiviral defense, and apoptosis. In erythroid precursors, IFN-α signaling has been shown to suppress ineffective erythropoiesis, enhance terminal maturation, and potentially alter the epigenetic landscape via JAK-STAT activation and histone-modifying enzymes [17–19]. These effects may help shift erythroid precursors toward more functional maturation in CDA-I patients.
While ropeginterferon alfa-2b has demonstrated a favorable safety profile in myeloproliferative neoplasms, potential risks such as autoimmune manifestations (e.g., thyroiditis, lupus-like syndromes) [20] and abnormal liver function[21] have been reported and warrant close monitoring. In our patients, no significant immune-related adverse events or hepatotoxicity were observed during follow-up.
Notably, with respect to dosing considerations, the study by Miyachi et al. reported that systemic exposure to ropeginterferon alfa-2b in Asian individuals was approximately 1.7- to 2.0-fold greater than that in Caucasian subjects [22]. Although the incidence of adverse events did not significantly differ between the two populations, these pharmacokinetic differences suggest that a more conservative dose-escalation strategy may be appropriate for Asian patients.
Our report is the first to show that ropeginterferon alfa-2b remained effective in two patients with CDA-I but enhanced their quality of life by reducing the burden of injections. Both patients achieved optimal disease control with a biweekly dose of 250 µg, achieved normal hemoglobin levels and improved hyperbilirubinemia. Given the rarity of CDA-I, this study highlights the potential of pegylated interferon as a viable long-term treatment strategy. In summary, we present the first documented use of ropeginterferon alfa-2b in CDA-I patients, demonstrating its efficacy with less frequent dosing and better tolerability than traditional IFN-α. These cases underscore the role of genetic testing in CDA-I diagnosis and highlight pegylated interferon as a potential long-term treatment option. Further research is needed to validate its broader clinical utility.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Babbs C, Roberts NA, Sanchez-Pulido L, Mc Gowan SJ, Ahmed MR, Brown JM, Sabry MA, Consortium WGS, Bentley DR, Mc Vean GA, Donnelly P, Gileadi O, Ponting CP, Higgs DR, Buckle VJ. Homozygous mutations in a predicted endonuclease are a novel cause of congenital dyserythropoietic anemia type I. Haematologica. 2013;98(9):1383–7. 10.3324/haematol.2013.089490.10.3324/haematol.2013.089490 PMC 376209423716552 · doi ↗ · pubmed ↗
- 2Pockros PJ, Carithers R, Desmond P, Dhumeaux D, Fried MW, Marcellin P, Shiffman ML, Minuk G, Reddy KR, Reindollar RW, Lin A, Brunda MJ, Group PIS. Efficacy and safety of two-dose regimens of peginterferon alpha-2a compared with interferon alpha-2a in chronic hepatitis C: a multicenter, randomized controlled trial. Am J Gastroenterol. 2004;99(7):1298–305. 10.1111/j.1572-0241.2004.30306.x.10.1111/j.1572-0241.2004.30306.x 15233669 · doi ↗ · pubmed ↗