De Novo Splice Site Variant of TCF12 in a Boy With Isolated Kallmann Syndrome
Erina Suzuki, Hirohito Shima, Aki Ueda, Kazuhiko Nakabayashi, Keiko Matsubara, Yoko Kuroki, Junko Kanno, Maki Fukami

TL;DR
A de novo mutation in the TCF12 gene is linked to isolated Kallmann syndrome in a boy with no other symptoms.
Contribution
This is the first report of a TCF12 splice site variant causing isolated Kallmann syndrome.
Findings
A de novo TCF12 splice site variant (c.391–1G >A) was identified in a patient with Kallmann syndrome.
The variant was predicted to cause a frameshift and premature termination, and was classified as pathogenic.
No other rare variants in known Kallmann syndrome or CHH genes were found in the patient.
Abstract
Background: Kallmann syndrome is a rare endocrinopathy characterized by congenital hypogonadotropic hypogonadism (CHH) and olfactory dysfunction. The current understanding of the genetic basis of Kallmann syndrome is fragmentary. TCF12 is a causative gene for autosomal dominant craniosynostosis with various complications. Although recent studies identified rare nucleotide substitutions and indels of TCF12 in a few families with CHH and additional clinical features, the significance of TCF12 variants as the cause of Kallmann syndrome remains uncertain. Case Presentation: A Japanese boy exhibited bilateral cryptorchidism and micropenis at birth. He was otherwise healthy and had normal developmental milestones. At 11 years of age, he showed no pubertal signs. Physical examinations detected no clinical abnormalities except hyposmia. Brain imaging suggested olfactory bulb hypoplasia, but no…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
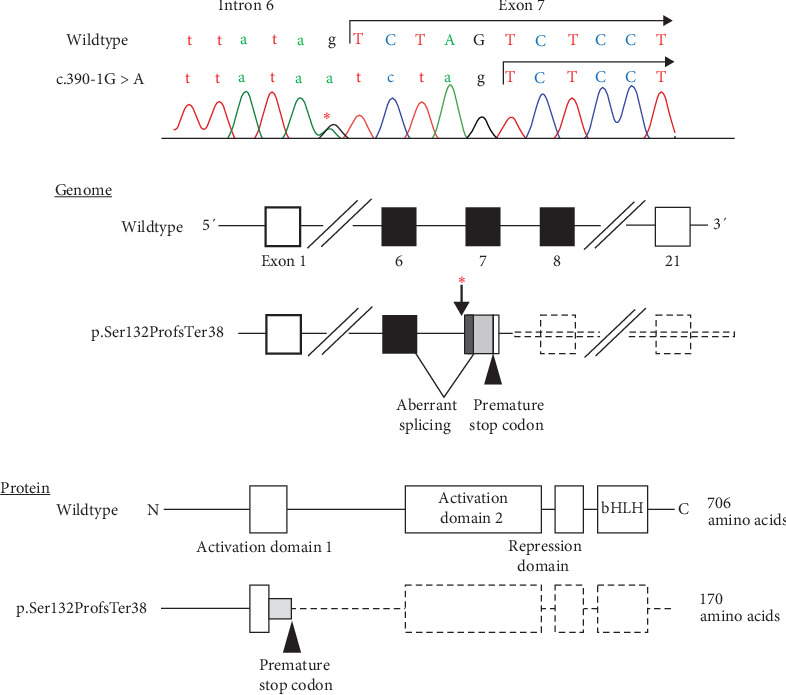 Figure 1
Figure 1- —Japan Agency for Medical Research and Development
- —National Center for Child Health and Development
- —Japan Endocrine Society
- —Canon Foundation
- —Takeda Science Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and Clinical Aspects of Sex Determination and Chromosomal Abnormalities · Genetics and Neurodevelopmental Disorders · Hypothalamic control of reproductive hormones
1. Introduction
Kallmann syndrome is a rare endocrine disorder characterized by congenital hypogonadotropic hypogonadism (CHH) and olfactory dysfunction [1]. Kallmann syndrome results from monoallelic, biallelic, or oligogenic mutations in genes involved in hormone secretion from the hypothalamus or pituitary [1]. To date, more than 30 genes have been implicated in Kallmann syndrome or normosmic CHH, although pathogenic variants in these genes can explain less than half of the cases [1].
TCF12 (NM_207037.2, alias HTF4) is a ubiquitously expressed gene that encodes a member of the basic helix-loop-helix (bHLH) transcription factor. TCF12 is known as a causative gene for autosomal dominant craniosynostosis [2]. Previous studies have suggested that TCF12 heterodimerizes with TWIST1 and regulates specification of the boundary between neural crest and cephalic mesoderm [2]. Patients with heterozygous TCF12 variants frequently exhibit skeletal malformations, facial dysmorphisms, and intellectual problems, in addition to craniosynostosis [3]. Some of these patients were reported to have CHH [3]. In 2020, Davis et al. [4] identified rare nucleotide substitutions and indels of TCF12 in 13 families with CHH. Most variant-positive individuals exhibited Kallmann syndrome with dental, neurobehavioral, or musculoskeletal abnormalities. Subsequently, Celik et al. [5] have detected intragenic deletions of TCF12 in a patient with Kallmann syndrome and thyroid tumor. These findings imply that TCF12 is a candidate gene for Kallmann syndrome. However, there are no further reports of TCF12 variants in patients with Kallmann syndrome. Since most patients reported by Davis et al. and Celik et al. [4, 5] exhibited complex phenotypes, the etiological relationship between TCF12 variants and isolated Kallmann syndrome remains unknown. Here, we report a boy with a novel de novo splice site variant of TCF12 and isolated Kallmann syndrome.
2. Case Presentation
The case was a Japanese boy born to a healthy, unrelated couple. At birth, he was noted to have bilateral cryptorchidism and micropenis. Endocrine evaluation at 1 month of age during minipuberty showed low levels of luteinizing hormone (LH), follicle-stimulating hormone (FSH), and testosterone (Table 1). Thus, he was suspected of having CHH and received testosterone injections and orchidopexy during infancy. He was otherwise healthy and showed normal development and growth. At 11 years and 4 months of age, he visited our hospital for clinical evaluation. His height and weight were 141 cm (−0.47 SD) and 45.4 kg (+0.90 SD). He could detect strong odors, but did not recognize any odors of foods or other items in daily life. He had no visual or hearing impairment. Physical examinations detected no signs of craniofacial, neurobehavioral, or musculoskeletal abnormalities. He showed prepubertal external genitalia of Tanner stage I. Brain imaging suggested hypoplasia of the olfactory bulbs, but no other anomalies. He did not undergo further imaging examinations. Endocrine examinations revealed low gonadotropin responses to gonadotropin-releasing hormone (GnRH) stimulation, together with normal blood levels of other hormones examined (Table 1). He was clinically diagnosed with isolated Kallmann syndrome.
The patient was identified through mutation screening of TCF12 for patients with etiology-unknown CHH. We performed whole-exome sequencing (WES) for eight patients with Kallmann syndrome or normosmic CHH who carried no mutations in 14 major causative genes for CHH (ANOS1, CHD7, FGF8, FGFR1, GNRH1, GNRHR, KISS1, KISS1R, PROK2, PROKR2, SOX10, TAC3, TACR3, and WDR11). We searched for rare protein-altering variants in 57 genes reported as causative/candidate genes for CHH or Kallmann syndrome (Table S1) [6, 7]. The methods of WES and variant calling were reported previously [8]. As a result, we identified a rare TCF12 variant in one patient (the present case). The patient carried a heterozygous c.391–1G > A variant, which affected a consensus nucleotide at a splice acceptor site of exon 7. The presence of the variant was confirmed by Sanger sequencing (Figure 1A). In silico prediction using SpliceAI (https://spliceailookup.broadinstitute.org/) suggested that the variant likely decreased the activity of the original splice site and instead activated a nearby cryptic splice acceptor site in exon 7. Hence, this variant was assumed to intronize the first five nucleotides of exon 7 to cause a frameshift from the 132nd codon and resultant premature termination at the 170th codon (p.Ser132ProfsTer38) (Figure 1B). The variant was not found in the Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org/) and Tohoku Medical Megabank Organization Database (ToMMo, https://www.megabank.tohoku.ac.jp/). The variant was not identified in the parents of the patient. The variant was assessed as pathogenic (PVS1 + PM2 + PS2) according to the ACMG/AMP 2015 guidelines [9]. WES identified no rare variants in the remaining 56 genes.
3. Discussion
We identified a hitherto unreported de novo TCF12 variant in a boy with Kallmann syndrome. The variant was assumed to activate a cryptic acceptor site in exon 7, leading to the intronization of the first five nucleotides of this exon. The mutant mRNA satisfies the condition of nonsense-mediated mRNA decay [10], and therefore, is likely to undergo rapid degradation. Even if the variant mRNA escapes nonsense-mediated decay, it would encode a truncating protein lacking the activation domain 2, the repression domain, and the bHLH domain (Figure 1C). Thus, this variant is likely to result in TCF12 haploinsufficiency. These results broaden the mutation spectrum of TCF12.
The patient exhibited typical clinical features of Kallmann syndrome but lacked other craniofacial, neurobehavioral, or musculoskeletal abnormalities. Thus far, pathogenic TCF12 variants have been identified mostly in patients with craniosynostosis or other developmental defects [2, 3]. Only two patients with frameshift or nonsense variants in TCF12 (individuals 4 and 7 in [4]) were reported to have Kallmann syndrome without other complications. The present study provides evidence that heterozygous loss-of-function variants of TCF12 are causes of isolated Kallmann syndrome. Since our patient carried no pathogenic variants in other known CHH-causative genes, monoallelic TCF12 variants appear to be sufficient to cause Kallmann syndrome as a Mendelian disorder. Consistent with this, animal studies have shown that Tcf12 is abundantly expressed in the embryonic ectoderm and neural folds, and loss of Tcf12 perturbs GnRH neuronal patterning in zebrafish [4]. Since TCF12 is a transcription factor [2], it may transactivate some genes involved in the development of GnRH neurons. In this context, Davies et al. [4] have proposed that TCF12 interacts with other GnRH neuron-associated molecules such as STUB1 and SOX10. The underlying mechanisms of Kallmann syndrome in TCF12-mutation-positive patients need to be clarified in future studies.
Two matters are noteworthy for the TCF12 variant in the present case. First, the patient was identified through WES for eight patients with etiology-unknown CHH. These results, in conjunction with previous findings that only 13 of 729 pedigrees with Kallmann syndrome carried TCF12 variants [4], indicate that TCF12 abnormalities account for only a small percentage of the etiology of Kallmann syndrome. Second, the patient lacked craniosynostosis or other malformations despite having a protein-truncating variant in TCF12. Thus far, no apparent genotype–phenotype correlation has been observed in TCF12 abnormalities [4]. Future studies are needed to clarify the phenotypic determinants of this condition.
We identified a novel de novo splice site variant of TCF12 in a boy with Kallmann syndrome without other clinical features. Our data broaden the mutation spectrum of TCF12. More importantly, this study argues for the etiological relationship between TCF12 variants and isolated Kallmann syndrome.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Young J. Xu C. Papadakis G. E. Clinical Management of Congenital Hypogonadotropic Hypogonadism Endocrine Reviews 201940266971010.1210/er.2018-001162-s 2.0-8506326649830698671 · doi ↗ · pubmed ↗
- 2Sharma V. P. Fenwick A. L. Brockop M. S. Mutations in TCF 12, Encoding a Basic Helix-Loop-Helix Partner of TWIST 1, are a Frequent Cause of Coronal Craniosynostosis Nature Genetics 201345330430710.1038/ng.25312-s 2.0-8487462687723354436 PMC 3647333 · doi ↗ · pubmed ↗
- 3Paumard-Hernández B. Berges-Soria J. Barroso E. Expanding the Mutation Spectrum in 182 Spanish Probands With Craniosynostosis: Identification and Characterization of Novel TCF 12 Variants European Journal of Human Genetics 201523790791410.1038/ejhg.2014.2052-s 2.0-8493087193025271085 PMC 4463497 · doi ↗ · pubmed ↗
- 4Davis E. E. Balasubramanian R. Kupchinsky Z. A. TCF 12 Haploinsufficiency Causes Autosomal Dominant Kallmann Syndrome and Reveals Network-Level Interactions Between Causal Loci Human Molecular Genetics 202029142435245010.1093/hmg/ddaa 12032620954 PMC 7608740 · doi ↗ · pubmed ↗
- 5Celik N. B. Sezer A. Genel N. Savas-Erdeve S. Karamanİ. Cetinkaya S. Case Report: An Adolescent Female With Anosmic Hypogonadotropic Hypogonadism, Intellectual Disability, and Papillary Thyroid Carcinoma: Heterozygous Deletion of TCF 12 Frontiers in Endocrinology 2024151510.3389/fendo.2024.14269161426916 PMC 1125791239036055 · doi ↗ · pubmed ↗
- 6Oleari R. Massa V. Cariboni A. Lettieri A. The Differential Roles for Neurodevelopmental and Neuroendocrine Genes in Shaping Gn RH Neuron Physiology and Deficiency International Journal of Molecular Sciences 2021221710.3390/ijms 221794259425 PMC 843160734502334 · doi ↗ · pubmed ↗
- 7Sayed Y. Al Howard S. R. Panel Testing for the Molecular Genetic Diagnosis of Congenital Hypogonadotropic Hypogonadism – A Clinical Perspective European Journal of Human Genetics 202331438739410.1038/s 41431-022-01261-036517585 PMC 10133250 · doi ↗ · pubmed ↗
- 8Kinjo K. Nagasaki K. Muroya K. Rare Variant of the Epigenetic Regulator SMCHD 1 in a Patient With Pituitary Hormone Deficiency Scientific Reports 202010110.1038/s 41598-020-67715-x 10985 PMC 733516132620854 · doi ↗ · pubmed ↗