Genome assembly of Bougainvillia cf. muscus (Cnidaria: Hydrozoa)
Aide Macias-Muñoz, Rebecca Varney, Eva Katcher, Maia Everhart, Todd H Oakley

TL;DR
This paper presents a high-quality genome assembly of Bougainvillia cf. muscus, a hydrozoan with simple eyes, offering insights into vision-related genes and genome evolution in Cnidaria.
Contribution
The study provides the first genome assembly of an eyed hydrozoan species, enabling comparative genomic analyses of vision-related genes and synteny.
Findings
The Bougainvillia genome shows high macrosynteny with other hydrozoans like Hydra vulgaris and Hydractinia symbiolongicarpus.
Repetitive elements constitute 62% of the Bougainvillia genome.
Twenty cnidarian opsins were identified, with evidence of gene duplication and loss in vision-related gene families.
Abstract
As one of just a handful of nonbilaterian animal phyla, Cnidaria are key to understanding genome evolution across Metazoa. Despite their importance and diversity, the genomes of most species in the phylum are unsequenced, due in large part to difficulties cultivating them in a laboratory. Here, we present a genome sequence of Bougainvillia cf. muscus, a hydrozoan with 4 marginal bulbs each containing 7 simple eyes (ocelli). This species appeared in our tanks from contamination. While we lacked sufficient samples for transcriptomic or functional studies, we were able to expand our knowledge of how the genome of this species compares to the few, better studied members of hydrozoans by investigating synteny to other cnidarians, repetitive element content, and phylogenetics and synteny of vision-related genes in this eyed species compared to eyeless relatives. The genome sequence consists…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
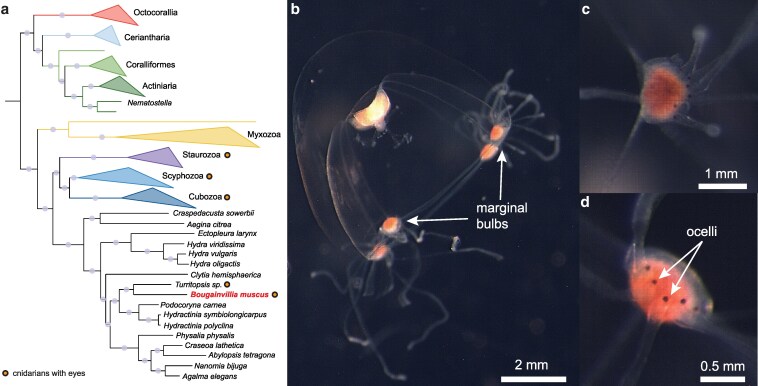 Fig. 1
Fig. 1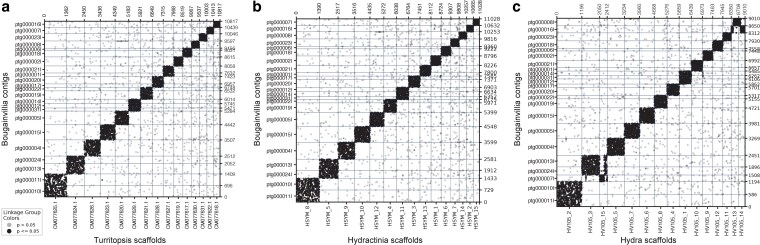 Fig. 2
Fig. 2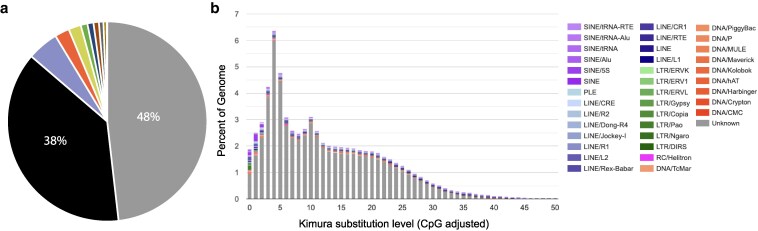 Fig. 3
Fig. 3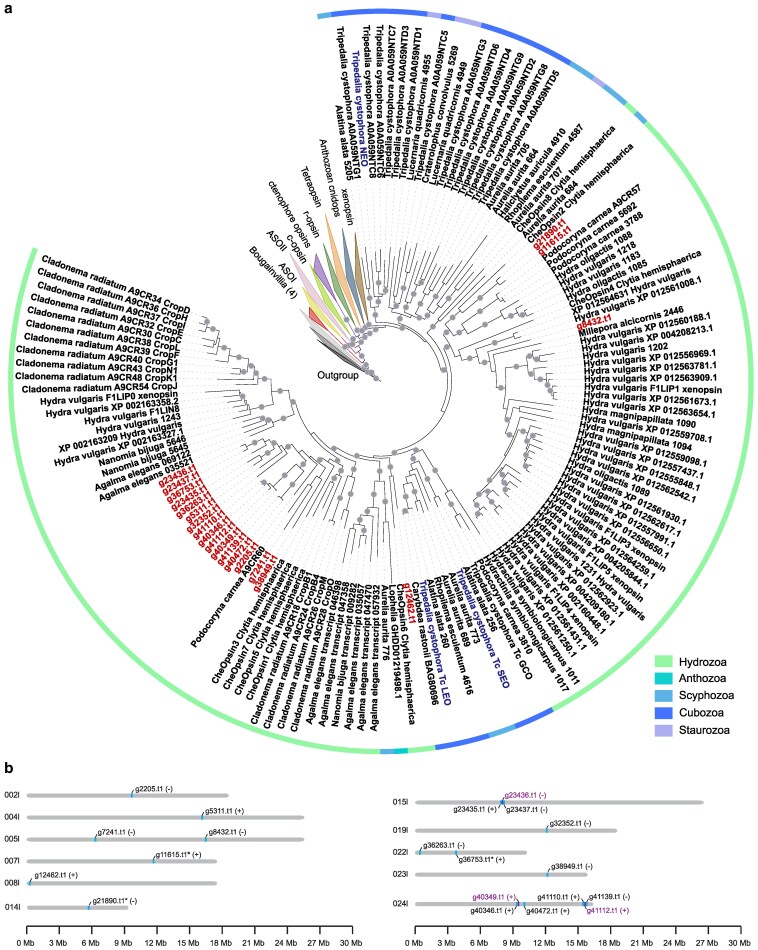 Fig. 4
Fig. 4| Genome feature | Value |
|---|---|
| Cell 1 bases read | 474.02 Gb |
| Cell 1 polymerase reads | 4,472,747 |
| Cell 2 bases read | 438.52 Gb |
| Cell 2 polymerase reads | 4,076,913 |
| Genome length | 375.33 Mb |
| Contig number | 350 |
| Contig N50 | 10 Mb |
| Contig L50 | 16.62 Mb |
| Contig N90 | 22 Mb |
| Contig L90 | 6.465 Mb |
| Max contig length | 26.35 Mb |
| GC | 36.21% |
| BUSCO | 90.10% |
| Genes | 46,431 |
| Number of elements | Length occupied (bp) | Percentage of sequence | |
|---|---|---|---|
| Retroelements | 87,012 | 26,807,637 | 7.14 |
| SINEs | 12,334 | 4,105,380 | 1.09 |
| Penelope | 10,802 | 3,305,874 | 0.88 |
| LINEs | 65,407 | 19,011,721 | 5.07 |
| CRE/SLACS | 748 | 360,091 | 0.1 |
| L2/CR1/Rex | 18,383 | 4,904,952 | 1.31 |
| R1/LOA/Jockey | 347 | 47,488 | 0.01 |
| R2/R4/NeSL | 997 | 714,097 | 0.19 |
| RTE/Bov-B | 14,312 | 4,773,644 | 1.27 |
| L1/CIN4 | 1,110 | 216,104 | 0.06 |
| LTR elements | 9,271 | 3,690,536 | 0.98 |
| BEL/Pao | 1,448 | 1,148,511 | 0.31 |
| Ty1/Copia | 160 | 124,659 | 0.03 |
| Gypsy/DIRS1 | 3,420 | 1,781,405 | 0.47 |
| Retroviral | 3,792 | 352,487 | 0.09 |
| DNA transposons | 102,744 | 8,895,639 | 2.37 |
| hobo-Activator | 2,781 | 353,672 | 0.09 |
| Tc1-IS630-Pogo | 3,510 | 437,421 | 0.12 |
| En-Spm | — | — | 0 |
| MULE-MuDR | 211 | 32,456 | 0.01 |
| PiggyBac | 51 | 28,969 | 0.01 |
| Tourist/harbinger | 1,042 | 264,753 | 0.07 |
| Other | 12,370 | 870,658 | 0.23 |
| Rolling circles | 17,411 | 2,737,491 | 0.73 |
| Unclassified | 1,322,062 | 181,344,581 | 48.32 |
| Total interspersed repeats | 220,353,731 | 58.71 | |
| Small RNA | 7,753 | 7,589,974 | 2.02 |
| Satellites | — | — | 0 |
| Simple repeats | 41,673 | 2,003,055 | 0.53 |
| Low complexity | 6,601 | 306,921 | 0.08 |
| Total bases masked | — | 231,870,804 | 61.78 |
|
|
|
| ||
|---|---|---|---|---|
| Visual system specification | ||||
| BMP/GDF | 7 | 9 | 8 |
|
| Hedgehog (Hh) | 3 | 1 | 2 |
|
| Wnt | 10 | 11 | 12 |
|
| Retinal determination | ||||
| Eyes absent | 1 | 1 | 1 |
|
| Pax | 3 | 3 | 3 |
|
| SIX | 2 | 4 | 4 |
|
| Phototransduction | ||||
| Galphai | 1 | 1 | 1 |
|
| Galphas | 1 | 2 | 2 |
|
| Galphaq | 1 | 1 | 1 |
|
| Gbeta | 3 | 2 | 3 |
|
| CNG | 4 | 2 | 3 |
|
| TRPC | 1 | 1 | 1 |
|
| PLC | 6 | 2 | 1 |
|
| AC13E | 2 | 1 | 1 |
|
| AC2/5-like | 2 | 2 | 2 |
|
| GC | 3 | 7 | 4 |
|
| Arrestin | 1 | 1 | 1 |
|
| Visual cycling | ||||
| GRK | 1 | 1 | 1 |
|
| Rhk | 1 | 1 | 1 |
|
| SEC14 | 2 | 3 | 5 |
|
| Phosphodiesterase | 5 | 3 | 3 |
|
- —NSF10.13039/100000001
- —American Philosophical Society Franklin Research
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMarine Invertebrate Physiology and Ecology · Marine Ecology and Invasive Species · Coral and Marine Ecosystems Studies
Introduction
Comparative genomic studies are essential for understanding biodiversity, phylogenetic relationships, genome evolution, and the origins of organismal traits. With a growing number of high-quality genome assemblies, researchers can now examine how genomic changes contribute to animal evolution. For example, synteny analyses of ctenophore and sponge genomes have clarified deep animal relationships (Schultz et al. 2023), and similar methods have linked genomic changes to key events in vertebrate and annelid evolution (Simakov et al. 2020; Lewin et al. 2024). These studies demonstrate the power of new genomes to inform questions about evolution.
An especially important group for genomic comparisons is the phylum Cnidaria—which includes jellyfish, corals, and sea anemones. With ∼13,300 described species and as a sister group to Bilateria, cnidarians are central to understanding early animal evolution. Although recent efforts have expanded available cnidarian genomes (Santander et al. 2022), most genomic resources are from model organisms such as Nematostella, Hydra, and Hydractinia (Chapman et al. 2010; Kon-Nanjo et al. 2023; Zimmermann et al. 2023). As a result, much of the group's morphological and ecological diversity remains poorly sampled at the genomic level. Increasing the taxonomic breadth of cnidarian genomics will help reveal gene-level differences underlying their diverse forms and functions (Travert et al. 2023).
Cnidarians are also particularly valuable for studying the evolution of visual systems. Eyes evolved convergently at least 9 times within the phylum (Picciani et al. 2018; Miranda and Collins 2019). Some cnidarian eyes share developmental gene families with those of bilaterians, although often using different orthologs (Plachetzki et al. 2007; Koyanagi et al. 2008; Suga et al. 2008; Vöcking et al. 2022). One species, the box jellyfish Tripedalia cystophora, is the subject of diverse visual system research with published studies on morphology, gene expression, and visually guided behaviors (Nilsson et al. 2005; Garm et al. 2007; Garm, Oskarsson, and Nilsson 2011; Bielecki et al. 2014).
We intended to sequence the genome of T. cystophora when our culture unexpectedly produced small medusae that we sent for sequencing before discovering they were a contaminant of the hydrozoan Bougainvillia cf. muscus (herein referred to as Bougainvillia). Although we were unable to obtain polyps or additional medusae of the contaminant, thus preventing further analyses, we successfully generated a high-quality genome sequence. Bougainvillia belongs to Filifera, a hydrozoan suborder with few genomic resources (Kayal et al. 2015). Within Filifera, eyes likely evolved multiple times convergently, with B. muscus representing a distinct eye origin (named eye origin 4a) compared to other Bougainvillia (4b), Turritopsis (4d), and other Filifera (origin 5) (Picciani et al. 2018). To date, research on Bougainvillia has focused on morphology and ecology, with no high-quality genomic resource available (but see Edsinger et al. 2024).
Here, we present the genome of an eyed Bougainvillia species (Fig. 1). This assembly enables comparisons of synteny among hydrozoans, phylogenetic placement within Cnidaria, and investigation of genes involved in eye development and function. This assembly represents the best genomic resource to date for Bougainvillia and adds critical data for understanding cnidarian visual system evolution and genome diversity.
B. cf. muscus falls within Hydrozoa and has ocelli. a) Cnidarian phylogeny using 748 orthologs from DeBiasse et al. (2024) with B. cf. muscus added. Branches that have over 80% bootstrap support are labeled with light gray circles. Circles adjacent to species or class names represent cnidarians that have eyes. b) Photograph of a Bougainvillia medusa with marginal bulbs labeled. c, d) Zoomed in images of the marginal bulbs showing the ocelli, which are rounded, black, and located at the base of each tentacle. Bougainvillia have 7 ocelli in each of their 4 marginal bulbs.
Materials and methods
DNA extraction
We obtained Bougainvillia from a tank of seawater at 25°C maintained at the University of California, Santa Barbara that also contained T. cystophora polyps. Water came from University of California, Santa Barbara's seawater system and the T. cystophora polyps came from a culture from Denmark, with T. cystophora originating from Puerto Rico or Florida. In September 2021, we began noticing many small medusae. We collected 15 free swimming medusae using plastic pipettes and washed them thoroughly with filtered seawater. We extracted DNA from fresh tissue using Circulomics Nanobind Tissue Big DNA Kit (NB-900-701-01; Circulomics via Pacific Biosciences, Menlo Park, CA, USA) skipping the tissueRuptor step.
Data processing
The University of California, Irvine Genomics High Throughput Facility made PacBio HiFi low input libraries from our extracted DNA and sequenced them on 2 SMRT cells using PacBio Sequel II. We processed HiFi reads from BAM files using ccs v 6.4.0. In cell 1, 2,085,669 (46.63%) zero-mode waveguides passed filter and 1,982,604 (48.63%) in cell 2. We used Bam2fastq v. 1.3.0 to combine the sequencing data from the 2 cells and to extract it as fastq files. We generated a genome assembly using hifiasm (Cheng et al. 2021) and then determined completeness and contiguity using BUSCO v. 5.3.2 -l metazoa and BBmap v. 38.96 (Bushnell et al. 2014; Simão et al. 2015). To check for potential contaminants, we used BlobToolKit v. 4.2.1 and removed outlier contigs with no overlap to Cnidaria and with GC content < 0.3 and > 0.5 (Challis et al. 2020). We used BRAKER3 (Gabriel et al. 2024) to annotate the final assembly using protein models from Hydra vulgaris (Chapman et al. 2010), Clytia hemisphaerica (Leclère et al. 2019), Hydractinia echinata, and Hydractinia symbiolongicarpus (Schnitzler et al. 2024) as input.
Species identification
We initially identified the medusae as B. muscus from morphological characters listed in the World Register of Marine Species (WoRMS Editorial Board 2024). After assembling the genome, we extracted sequences for Cytochrome c oxidase I (COI), 18S, 16S, and 28S ribosomal RNA genes and compared them to available sequences in NCBI via BLAST (Supplementary Table 1). The top hit was B. muscus. We then downloaded sequences of B. muscus COI, 18S, 16S, and 28S from NCBI and did reciprocal BLAST to the assembled genome. The sequences had an e-value of 0 suggesting a very close, if not exact match. For further validation, we downloaded COI, 16S, 18S, and 28S sequences for available Bougainvillia species from NCBI and generated phylogenetic trees using MAFFT to align sequences and iqtree2 with settings -m MFP -B 1000 -alrt 1000 -T 8 (Supplementary Fig. 1).
Cnidarian species phylogeny
We obtained sequences for 748 genes previously aligned across cnidarians from DeBiasse et al. (2024). We used hmm2aln.pl (https://github.com/josephryan/hmm2aln.pl) to identify sequences that matched each of these alignments in Bougainvillia. We then filtered for the best match (longest sequence with least gaps), ran MAFFT v. 7.520 (default parameters) and Gblocks v0.91b (-b2 = 10 -b3 = 10 -b4 = 5 -b5 = a), and concatenated the matrix (see ‘Data availability’). We generated a phylogenetic tree from the concatenated matrix using iqtree2 v. 2.2.2.6 -nt AUTO -bb 1000 -m TEST and then used iTOL v6 to visualize, root (outgroups Bilateria, Ctenophora, and Porifera), and annotate the tree. We also generated a tree from concatenated alignments prior to Gblocks using the same model.
Macrosynteny and repetitive sequences
To characterize synteny conservation across hydrozoans, we investigated macrosynteny between our final Bougainvillia genome assembly and the Turritopsis, Hydra, and Hydractinia chromosome-level genome assemblies (Simakov et al. 2020; Kon-Nanjo et al. 2023; Dong et al. 2024). We downloaded the Turritopsis rubra (GCA_039566895.2) and H. vulgaris v3 (GCF_022113875.1) genomes from NCBI and the H. symbiolongicarpus v2.0 (https://doi.org/10.6084/m9.figshare.22126232.v1) genome from Figshare (University of Vienna 2022; Kon-Nanjo et al. 2023; Yantai Institute of Coastal Zone Research 2024). We used odp with a minimum scaffold size of 1 Mb for the comparisons (Schultz et al. 2023). To estimate the degree of macrosynteny conservation, we calculated the number of one-to-one orthologs in homologous locations and divided by the total number of orthologs in the odp map (Wang et al. 2017; Li et al. 2022).
To identify repetitive sequences, we first generated a custom library using default parameters in RepeatModeler v. 2.0.5 (Flynn et al. 2020) and then used it to identify repeats in the genome assembly with RepeatMasker v. 4.1.5 (http://www.repeatmasker.org) (Smith et al. 2013-2015).
Vision-related phylogenetic trees
To investigate the phylogenetic relationships of vision-related genes across cnidarians, we downloaded publicly available transcriptomes from NCBI using NCBI's datasets command-line tools (Supplementary Table 2) (Sanders and Cartwright 2015; Chen et al. 2016; Hasegawa et al. 2016; Yunfeng et al. 2017; Lewis Ames et al. 2016; Kayal et al. 2018; Khalturin et al. 2019; Ying et al. 2019; Kupaeva et al. 2020; Xia et al. 2020; Ortiz González et al. 2021). We used TransDecoder v. 5.7.1 to extract coding and peptide sequences and cd-hit v. 4.8.1 with parameters -c 0.9 -n 5 to remove redundant transcripts (Li and Godzik 2006; Fu et al. 2012). We used OrthoFinder v. 2.5.5 to identify orthogroups among all datasets (Emms and Kelly 2019). We aligned sequences for candidate genes using MAFFT v. 7.520 and generated trees using iqtree2 v. 2.2.2.6 -m MFP -B 1000 -alrt 1000 -T 8 (Katoh and Standley 2013; Minh et al. 2020 ). We used iTOL v6 to annotate our final tree files, rooted at midpoint (Letunic and Bork 2024). For the opsin tree, we downloaded opsin sequences from (McCulloch et al. 2023) and searched for similar sequences in the Bougainvillia genome assembly using command-line BLAST+. We extracted sequences for the top hits and searched against the NCBI database to confirm opsin annotation. We added all Bougainvillia sequences that matched an opsin annotation to the opsin fasta file from (McCulloch et al. 2023) and used them as input for MAFFT and iqtree2. We rooted the tree using Trichoplax placopsins (Feuda et al. 2012; Fleming et al. 2020).
Results and discussion
B. cf. muscus morphology and phylogeny
Bougainvillia medusae were ∼6 mm in height and width and had 4 red structures at the corners of the bell. Higher magnifications revealed that these structures were tentacle bulbs, each with 7 simple eyes (ocelli) (Fig. 1). Medusae had a branching manubrium common to the genus (Fig. 1). Morphological identification coupled with 18S/COI/16S alignments and phylogenetic analyses all supported the species identification as B. muscus (Supplementary Fig. 1). A more data rich phylogenetic placement based on genes from across the genome was consistent with this identification, with Bougainvillia falling as sister to Turritopsis to form the Pseudothecata (Fig. 1; Supplementary Fig. 2) (Mendoza-Becerril et al. 2018).
Genome assembly
The final Bougainvillia genome assembly consisted of 350 contigs with an N50 of 10 Mb, max length of 26.349 Mb, and a total genome length of 375,328,287 bases (Table 1). Diverse cnidarian species including Hydra, Nematostella, Hydractinia, and Clytia have 15 chromosomes (Leclère et al. 2019; Simakov et al. 2022; Kon-Nanjo et al. 2023; Zimmermann et al. 2023), so we might expect a similar number in Bougainvillia. While we did not recover 15 chromosomes, the number of contigs is comparable to those of the Hydractinia genome before scaffolding with Hi-C (Kon-Nanjo et al. 2023). The Bougainvillia genome was smaller than those of other hydrozoans. The Hydractinia genome is ∼483 Mb, the Clytia genome is ∼445 Mb, the H. vulgaris 105 strain is ∼819 Mb, Hydra AEP is ∼901 Mb, and Hydra oligactis is ∼1,274 Mb (Leclère et al. 2019; Simakov et al. 2022; Cazet et al. 2023; Kon-Nanjo et al. 2023; Zimmermann et al. 2023). The GC content of the Bougainvillia genome assembly was 36.21%, indicating it is AT-rich like other hydrozoans (Chapman et al. 2010; Schnitzler et al. 2024).
The final Bougainvillia assembly had a complete BUSCO score of 90.1%. Using BRAKER3, we identified 46,431 predicted protein-coding genes (Table 1). This number is larger than those of the other cnidarian genomes, which have ∼20–30 thousand genes (Gold et al. 2019; Leclère et al. 2019; Cazet et al. 2023; Zimmermann et al. 2023). The number of predicted genes in our assembly is likely due to splice variants included as multiple separate gene models. To collapse similar sequences into single gene models, we clustered sequences using cd-hit at cutoff 0.9, yielding 33,515 predicted genes (BUSCO 90.8%). To our knowledge, this is the most contiguous assembly for a hydrozoan outside of Hydra, Hydractinia, and Turritopsis.
Synteny
Due to the limited number of high-quality genomes within Cnidaria, the degree of macrosynteny across the phylum remains understudied. However, previous studies in cnidaria suggest some phylogenetic signal in macrosynteny (Kon-Nanjo et al. 2023; Zimmermann et al. 2023). For example, Nematostella has a high degree of macrosynteny conservation with a closely related sea anemone (Scolanthus) but less conservation with Hydra and Xenia (Zimmermann et al. 2023). To determine the level of macrosynteny conservation between Bougainvillia and other hydrozoans, we compared our assembly to the chromosome-level assemblies of T. rubra, H. vulgaris 105 strain, and H. symbiolongicarpus. Hydra and Hydractinia have high macrosynteny conservation, with 2 potential translocations (Kon-Nanjo et al. 2023). Here, we found a high degree of macrosynteny conservation in Bougainvillia compared to Turritopsis and Hydractinia (Fig. 2). We did not detect any obvious genome rearrangements and calculated the conservation index to be 0.866 and 0.863, respectively.
Bougainvillia is syntenic with other hydrozoans. a) Oxford dot plots showing the location of 10,897 orthologous genes in 23 Bougainvillia contigs and 15 T. rubra scaffolds. b) Oxford dot plots showing the location of 11,029 orthologous genes in 23 Bougainvillia contigs and 15 H. symbiolongicarpus scaffolds. c) Oxford dot plots showing the location of 9,011 orthologous genes in 23 Bougainvillia contigs and 15 H. vulgaris scaffolds.
Between Bougainvillia and Hydra, we found macrosynteny to be mostly conserved, with some potential chromosome rearrangements (Fig. 2b). Firstly, there was a potential translocation between Hydra chromosomes 3 and 15 and Bougainvillia contigs ptg000013l, ptg000024l, and ptg000007l (Fig. 2). Bougainvillia contigs ptg000013l and ptg000024l were syntenic to most of Hydra chromosome 3 and part of chromosome 15. Meanwhile, ptg000007l was syntenic to a small part of Hydra chromosome 3 and syntenic to 2 pieces of chromosome 15. The other potential genomic rearrangement was ptg000016l, which was syntenic to parts of Hydra chromosomes 13 and 14. These translocations are consistent with the macrosynteny analysis between Hydra and Hydractinia supporting that the translocation occurred after the divergence of Hydra and the Hydractinia and Bougainvillia clade. The conservation index between Bougainvillia and Hydra was 0.838. Our results suggest that Bougainvillia is more syntenic with Turritopsis and Hydractinia than with Hydra, reflecting their phylogenetic relationships.
Repetitive elements
Repetitive elements make up ∼61% of the H. symbiolongicarpus genome and ∼70% of the H. vulgaris genome (Cazet et al. 2023; Kon-Nanjo et al. 2023). A recent study identified active TEs (Transposable elements) driving variation in genome size between 2 H. vulgaris strains (Kon-Nanjo et al. 2024). In addition, an in-depth characterization of TEs in Hydractinia found the subfamily Helitron is responsible for a recent expansion of repetitive elements (Kon et al. 2025). To compare repetitive content in Bougainvillia to other hydrozoans, we classified the number of repetitive elements in our genome assembly. Similar to Hydractinia, repetitive elements made up 61.78% of the Bougainvillia genome, with ∼48% being unclassified or unknown (Fig. 3a; Table 2). Following unclassified elements, retroelements in the LINE group made up the next largest percent of repetitive elements, 5% of the genome. Unlike in Hydractinia, whose sequence divergence analysis suggested 2 conspicuous episodes of repetitive element expansions, in Bougainvillia expansion of repetitive elements appears to be more continuous but also shows 2 peaks of repeat expansions (Fig. 3b).
The Bougainvillia genome is highly repetitive. a) Pie chart showing the percent of the genome made up of unclassified repetitive elements (gray), LINE (blue), DNA transposons (orange), small RNA (yellow), SINE (green), LTR (dark blue), Penelope (magenta), rolling circles (dark gray), simple repeats (brown), and unmasked (black). b) Repeat landscape of the Bougainvillia genome assembly.
Vision-related genes
Opsins
Opsin genes encode proteins that are typically used to detect light. Since Bougainvillia has ocelli, we examined opsin gene content. We identified a total of 24 predicted opsin-like genes in Bougainvillia (Fig. 4; Supplementary Table 3). On the opsin phylogenetic tree, we found 4 of the genes grouped outside of the cnidops (cnidarian opsin) group and were not opsins based on additional BLAST analyses, but rather other G protein-coupled receptors (Fig. 4). The remaining 20 genes were cnidops. This is comparable to the 17 opsins identified in Tripedalia (Liegertová et al. 2015). This number is expected for Medusozoa, which have a smaller diversity of opsins compared to Anthozoa (McCulloch et al. 2023).
Bougainvillia opsin phylogeny and genome location. a) Cnidarian opsin (cnidops) phylogeny. Bougainvillia opsins are labeled in red; they are the shortes genes that begin with the letter g and end with .t1. Tripedalia opsins known to be expressed in eyes are shown in blue. Branches that have over 80% bootstrap support are labeled with circles. b) Schematic of cnidops locations on the genome. Contigs are shown as gray bars, and cnidops locations are colored and labeled (typically blue). Genes that are located close to each other are labeled in purple. Orientation is shown after the gene name in parentheses. Genes with asterisks after their name are those that have introns.
Of the 20 cnidops genes, 2 group together on the phylogenetic tree (g21890.t1 and g11615.t1) and are closely related to Clytia opsin CheOpsin2. Another gene, g8432.t1, groups with a Hydra cnidops group. One Bougainvillia cnidops g12462.t1 groups together with CheOpsin6. The remaining cnidops form a clade together with a Podocoryna A9CR60 and Clytia CheOpsins 1, 5, 7, 3 (Fig. 4). In terms of phylogenetic position, none of the Bougainvillia opsin genes were orthologs of cnidops expressed in Tripedalia eyes (Bielecki et al. 2014; Garm et al. 2022) (Fig. 4). This suggests Bougainvillia may be using a nonorthologous opsin for visual function, indicating probable paralog switching. The 20 Bougainvillia cnidops genes are distributed across 11 chromosomes and all but 3 lack introns (Fig. 4; Supplementary Table 2). This is consistent with medusozoan opsins being intronless (Plachetzki et al. 2007; Liegertová et al. 2015). We also identified some cnidops that are located in tandem. This is the case for 3 Bougainvillia cnidops on contig ptg000015l and 2 pairs of genes on ptg000024l. Interestingly, the 6 cnidops genes on ptg000024l group together on the phylogenetic tree indicating similarity in gene sequence, including the 2 pairs of genes found in tandem (Fig. 4). The structure and location of the Bougainvillia opsins suggest molecular evolution by retrotransposition and tandem duplication similar to Hydra and Nematostella opsins (Macias-Munõz et al. 2019; McCulloch et al. 2023).
Other vision-related genes
To investigate the molecular evolution of other candidate vision-related genes in Bougainvillia, we identified genes similar to those involved in eye development, phototransduction, and visual cycling from model organisms. We generated phylogenetic trees and counted the number of genes present in each gene family within the Bougainvillia genome, T. cystophora (a box jellyfish, which all have eyes) transcriptome, and Hydra (an eyeless hydrozoan) gene models (Table 3; Supplementary Figs. 3–23). The gene families BMP/GDF, Hedgehog, and Wnt function in visual system specification and have undergone duplications and expansions in many animal groups, so we expected to find differences in copy number. We found several genes to be present in Bougainvillia and Tripedalia but absent in eyeless Hydra. These included dpp-like, wnt11-like, wnt4-like, and SIX1/2-like. Dpp—or decapentaplegic—is necessary for eye patterning in Drosophila and can induce ectopic expression of photoreceptor cell differentiation (Pignoni and Zipursky 1997). SIX-family genes also play a role in retinal specification in Drosophila. For phototransduction, G-alpha subunits, adenylate cyclase (AC), and cyclic nucleotide-gated (CNG) channels have been identified as probable components of cnidarian phototransduction cascades (Plachetzki et al. 2007; Koyanagi et al. 2008), but diversity across clades is possible (Vöcking et al. 2022). For phototransduction and visual cycling genes, we found some potential instances of gene duplication or loss across the 3 cnidarians. However, except for G-alpha-s (Koyanagi et al. 2008), the specific functions of these candidate genes in vision have never been demonstrated in any cnidarian. Future studies of eye development and phototransduction in cnidarians will reveal whether these genes play functional roles in eye patterning and light detection.
Conclusions
We generated a high-quality and contiguous genome for a hydrozoan with a visual system consisting of multiple ocelli. The Bougainvillia genome assembly consists of 350 contigs with an N50 of 10 Mb and a total genome length of 375 Mb. Although the Bougainvillia genome is smaller than other hydrozoans, macrosynteny is highly conserved with H. symbiolongicarpus and H. vulgaris, supporting synteny across hydrozoans. Similar to Hydractinia, ∼62% of the Bougainvillia genome was made up of repetitive elements. Exploration of vision-related genes identified candidate genes for future studies of eye development and light detection in cnidarians. This new hydrozoan genome is valuable for comparative studies in cnidarian and metazoan biology.
Supplementary Material
jkaf110_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bielecki J, Zaharoff AK, Leung NY, Garm A, Oakley TH. 2014. Ocular and extraocular expression of opsins in the rhopalium of Tripedalia cystophora (Cnidaria: Cubozoa). P Lo S One. 9(6):e 98870. doi:10.1371/journal.pone.0098870.24901369 PMC 4047050 · doi ↗ · pubmed ↗
- 2Bushnell B. et al 2014. ‘BB Map: a fast, accurate, splice-aware aligner. No. LBNL-7065 E.’ Lawrence Berkeley National Laboratory, Berkeley, CA. [Goo, September. 10.1186/1471-2105-13-238. · doi ↗
- 3Cazet JF, Siebert S, Little HM, Bertemes P, Primack AS, Ladurner P, Achrainer M, Fredriksen MT, Moreland RT, Singh S, et al 2023. A chromosome-scale epigenetic map of the Hydra genome reveals conserved regulators of cell state. Genome Res. 33(2):283–298. doi:10.1101/gr.277040.122.36639202 PMC 10069465 · doi ↗ · pubmed ↗
- 4Challis R, Richards E, Rajan J, Cochrane G, Blaxter M. 2020. Blob Tool Kit—interactive quality assessment of genome assemblies. G 3 (Bethesda). 10(4):1361–1374. doi:10.1534/g 3.119.400908.32071071 PMC 7144090 · doi ↗ · pubmed ↗
- 5Chapman JA, Kirkness EF, Simakov O, Hampson SE, Mitros T, Weinmaier T, Rattei T, Balasubramanian PG, Borman J, Busam D, et al 2010. The dynamic genome of Hydra. Nature. 464(7288):592–596. doi:10.1038/nature 08830.20228792 PMC 4479502 · doi ↗ · pubmed ↗
- 6Cheng H, Concepcion GT, Feng X, Zhang H, Li H. 2021. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 18(2):170–175. doi:10.1038/s 41592-020-01056-5.33526886 PMC 7961889 · doi ↗ · pubmed ↗
- 7De Biasse MB, Buckenmeyer A, Macrander J, Babonis LS, Bentlage B, Cartwright P, Prada C, Reitzel AM, Stampar SN, Collins AG, et al 2024. A cnidarian phylogenomic tree fitted with hundreds of 18S leaves. Bull Soc Syst Biol. 3(2). doi:10.18061/bssb.v 3i 2.9267. · doi ↗
- 8Dong Z, Wang F, Liu Y, Li Y, Yu H, Peng S, Sun T, Qu M, Sun K, Wang L, et al 2024. Genomic and single-cell analyses reveal genetic signatures of swimming pattern and diapause strategy in jellyfish. Nat Commun. 15(1):5936. doi:10.1038/s 41467-024-49848-z.39009560 PMC 11250803 · doi ↗ · pubmed ↗