Discovery of conformation constrained tetracyclic compounds as potent chitinase OfChi-h inhibitors with a novel binding mode
Pengtao Yuan, Xi Jiang, Xintong Ni, Xusheng Shao, Xuhong Qian, Qing Yang

TL;DR
Scientists discovered a new compound that strongly inhibits a key enzyme in insect molting, which could lead to better pest control methods.
Contribution
A novel conformationally constrained tetracyclic compound was developed as a potent inhibitor of lepidopteran chitinase with a unique binding mode.
Findings
Compound 6a inhibits OfChi-h with a Ki value of 58 nM.
Molecular docking and co-crystallization confirmed the compound's binding to three subsites of the enzyme.
The compound's structure offers a novel approach for developing insect chitinase inhibitors.
Abstract
Chitinase h (Chi-h) has been identified as a promising pesticide target due to its exclusive distribution in lepidopteran insects and its essential role in the moulting processes. In this study, we leverage OfChi-h from destructive agricultural pest Ostrinia furnacalis (Asian corn borer) as a model target to identify novel chitinase inhibitors. A conformational restriction approach was employed to design a series of novel OfChi-h inhibitors. Among these, compound 6a showed the highest inhibitory activity against OfChi-h, with a Ki value of 58 nM. Molecular docking analysis suggested that 6a tightly bound to three subsites (-3 to −1) of OfChi-h. The binding mode is further confirmed by the co-crystallization data of 6a with the SmChiA, a bacterial homologue of OfChi-h, at a resolution of 1.8 Å. This research presents a novel approach for the development of highly potent insect chitinase…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
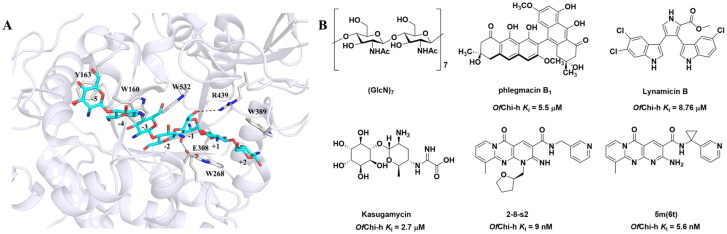 Figure 1
Figure 1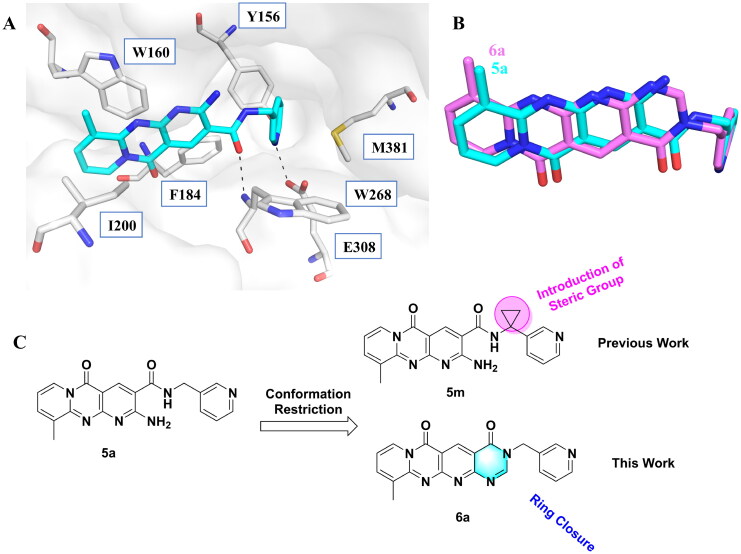 Figure 2
Figure 2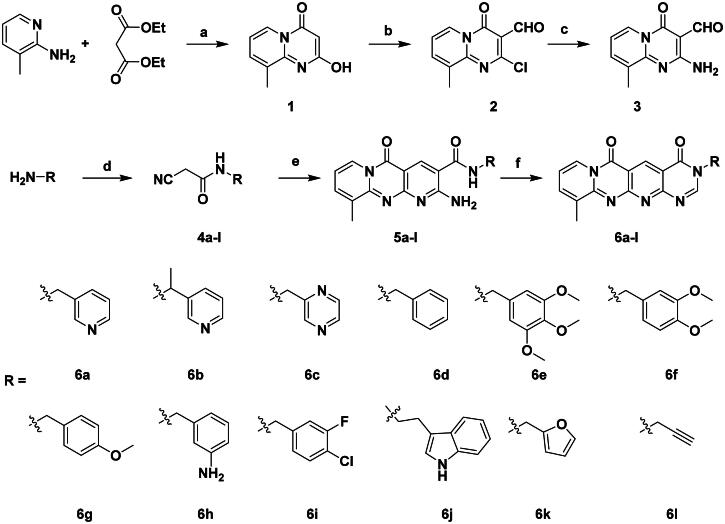 Scheme 1
Scheme 1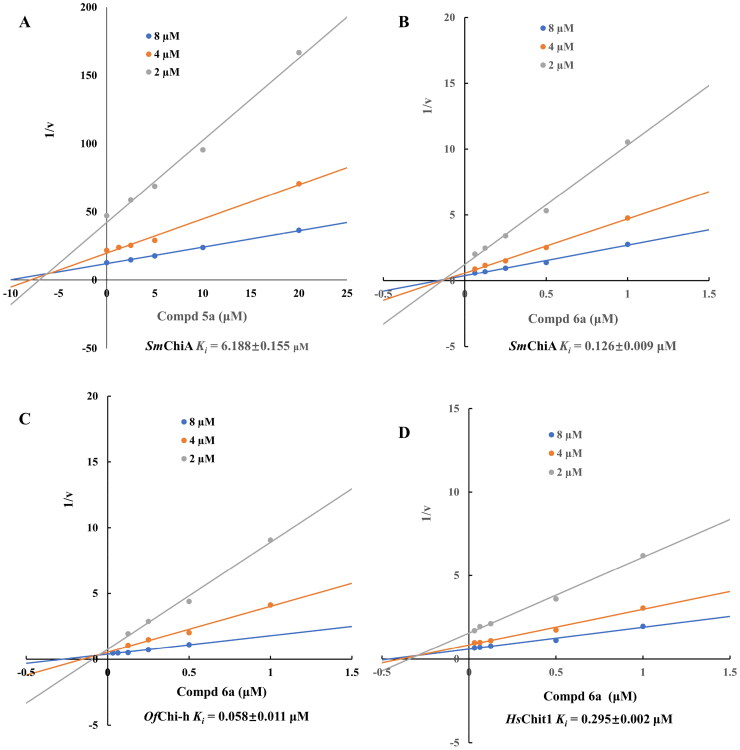 Figure 3
Figure 3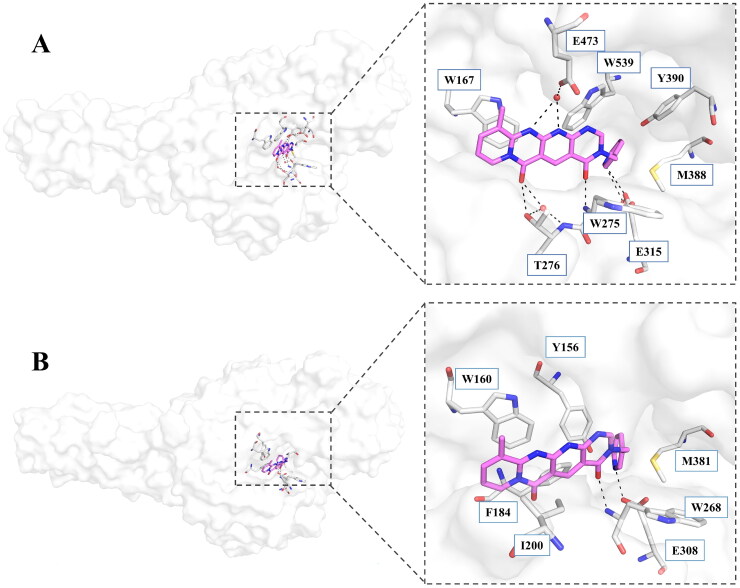 Figure 4
Figure 4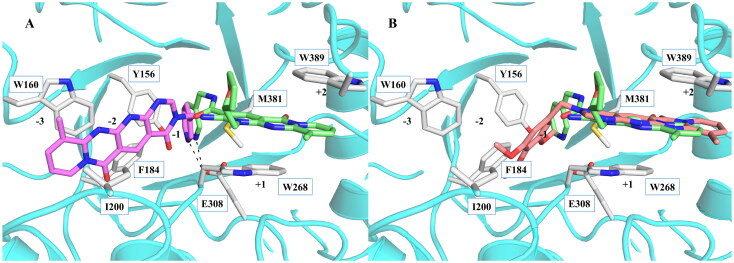 Figure 5
Figure 5|
| ||||
|---|---|---|---|---|
| Compd | R | |||
| 10 μM (%) | 10 μM (%) | |||
|
|
| 98.5 ± 0.2% | 99.3 ± 0.2% | 0.058 |
|
|
| 88.7 ± 2.2% | 96.8 ± 0.1% | 0.215 |
|
|
| 91.5 ± 0.8% | 95.3 ± 0.5% | 0.294 |
|
|
| 32.0 ± 1.4% | 72.5 ± 2.4% | 1.419 |
|
|
| 28.2 ± 4.8% | 96.2 ± 0.3% | 0.065 |
|
|
| 34.2 ± 2.5% | 69.3 ± 4.4% | 0.747 |
|
|
| 30.3 ± 2.7% | 74.0 ± 2.0% | 1.032 |
|
|
| 37.9 ± 0.4% | 89.0 ± 1.5% | 0.276 |
|
|
| 31.7 ± 0.2% | 42.9 ± 1.0% | 2.889 |
|
|
| 42.4 ± 4.1% | 49.6 ± 2.2% | 0.551 |
|
|
| 47.5 ± 1.2% | 86.8 ± 0.8% | 1.509 |
|
|
| 37.5 ± 2.2% | 81.0 ± 0.8% | 1.338 |
|
|
| – | – | 0.86 |
| Protein Data Bank entry | 7FD6 |
| Space group | C2221 |
| Unit-cell parameters | |
| a (Å) | 132.525 |
| b (Å) | 202.186 |
| c (Å) | 59.43 |
| α (°) | 90 |
| β (°) | 90 |
| γ (°) | 90 |
| Wavelength (Å) | 0.97776 |
| Temperature (K) | 100 |
| Resolution (Å) | 34.92–1.786 (1.85–1.786) |
| Unique reflections | 74412 |
| Observed reflections | 117961 |
| | 0.03271 (0.01386) |
| Average multiplicity | 1.6 (1.0) |
| <σ(I)> | 14.76 (3.08) |
| Completeness (%) | 98 (82) |
| | 0.1748/ 0.1917 |
| Protein atoms | 4124 |
| Water molecules | 678 |
| Other atoms | 28 |
| R.M.S. deviation from ideal | |
| Bond lengths (Å) | 0.004 |
| Bond angles (°) | 0.682 |
| Wilson B factor (Å2) | 18.57 |
| Average B factor (Å2) | 23.32 |
| Protein atoms | 21.64 |
| Water molecules | 33.20 |
| Ligand molecules | 31.57 |
| Ramachandran plot (%) | |
| Favored | 99 |
| Allowed | 1.5 |
| Outliers | 0 |
- —National Key Research and Development Program of China10.13039/501100012166
- —Shenzhen Science and Technology Program
- —Key Project of the Shanghai Science and Technology Committee
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsStudies on Chitinases and Chitosanases · Sulfur-Based Synthesis Techniques · Peptidase Inhibition and Analysis
Introduction
Chitinase (EC 3.2.1.14) is an enzyme that hydrolyses β-1,4-glycosidic bonds in chitin and chitooligosaccharides. The loss of chitinase enzymatic activity in insects results in severe exoskeletal defects and lethality at all developmental stages, as chitin is an important structural component of insect cuticle1. Chitinase-h(Chi-h) is a lepidoptera-exclusive insect chitinase that is absent in most beneficial insects including parasitic wasps and bees. Chi-h is thought to be acquired through horizontal gene transfer from bacteria2 and it shows significantly lower sequence similarity to other insect chitinases. Moreover, the structure of Chi-h is significantly different from that of human and other insect chitinases3. Thus, Chi-h is a promising target, and its inhibitors hold potential for eco-friendly pesticide development, contributing to sustainable agriculture4.
The only crystal structure of Chi-h is derived from Ostrinia furnacalis (OfChi-h), thereby establishing it as an important model for the development of Chi-h inhibitors1^,^3–5. OfChi-h features an elongated, asymmetric substrate-binding cleft containing seven distinct sugar-binding subsites, with multiple aromatic residues strategically positioned along the groove’s architecture. The co-crystal complex of OfChi-h**-(GlcN)7 revealed that seven subsites, spanning from −5 to +2, are present in the binding cleft3 (Figure 1(a)). The sugar-binding subsites were named according to Davies et al.6 where subsite -n represents the non-reducing end, subsite + n represents the reducing end, and enzymatic cleavage occurs between the −1 and the +1 subsites. To date, several OfChi-h inhibitors have been reported, including chitoheptaose ((GlcN)7**)3, the microbial secondary metabolite phlegmacin B17, the marine natural product Lynamicin B8, the aminoglycoside antibiotic Kasugamycin9, compound 2–8-s210, compound 5 m (also named as 6t in our previous report)11 and berberine along with its derivatives12^,^13. Several multitarget inhibitors of insect chitinolytic enzymes have also been reported, including Shikonin, Wogonin14, 3,5-Di-O-caffeoylquinic acid and γ-mangostin15 Rhein and its dimeric analogue Sennidin B16, Butenolide derivatives17^,^18 and guanidine-based inhibitors19–22.
Binding clef of OfChi-h and its inhibitors. (a) OfChi-h in complex with (GlcN)7 (PDB: 5GQB) and (b) representative chitinase OfChi-h inhibitors.
Conformational restriction is a well-known strategy in drug and pesticide development23^,^24, which could be achieved by the enhancement of steric effects25, intramolecular hydrogen bonding26, ion pairing27, π–π stacking, incorporation of cyclic elements28, or fusion of adjacent rings. Compared with flexible ligands, similarly restrained ligands can form analogous pairwise interactions with the target while sacrificing fewer conformational degrees of freedom during complex formation, thereby resulting in a lower entropic penalty29 and improved binding affinity. In addition, conformational restriction by introducing fused rings offers potential advantages, such as reducing degradation and increasing selectivity towards certain receptor subtypes30.
GH18 chitinases possess surface-exposed aromatic residues in the binding cleft, which interact with sugars by a combination of hydrogen bonding and hydrophobic stacking interactions and have been shown to be essential for substrate binding. Although all the GH18 chitinases use the same catalytic mechanism, they exhibit variations in the shape of their substrate-binding cleft. Thus, rigid structures with fused rings may selectively inhibit certain types of chitinases. In our previous study, we identified 5 m11 as a potent OfChi-h inhibitor by introducing steric groups to restrain the conformation of 5a (Figure 1(b)). In this work, we reported the discovery of a novel rigid tetracyclic scaffold for OfChi-h inhibitors by ring closure to fix the conformation, with the expectation that it will enhance both inhibitory activity and selectivity. Among the designed compounds, compound 6a showed the highest inhibitory activity against OfChi-h with a K_i_ value of 58 nM. Furthermore, the structural analysis and docking results revealed that the tetracyclic scaffold binds to different sites within the chitinase, which will guide future chitinase inhibitors design.
Results and discussion
Rational design of a novel tetracyclic core
As previously reported, compound 5a was involved in several key interactions with residues in the active site of the chitinase as revealed by its X-ray cocrystal structure11. For example, the pyridopyrimidine moiety of compound 5a stacked with Trp97 and Trp220 at +1 and +2 subsites, respectively, and the 3-pyridyl nitrogen formed a hydrogen bond with the key catalytic residue Glu144. Interestingly, molecular docking indicated that 5a binds to OfChi-h in a different way. It is predicted to bind in the −3, −2 and −1 subsites of OfChi-h, while still forming a hydrogen bond with the key catalytic residue Glu308 (Figure 2(a)). In addition, the carbonyl oxygen atom of the amide group forms a hydrogen bond with the backbone of Trp268. Therefore, we designed a tetracyclic scaffold, compound 6a, with fewer rotatable bonds to lock it covalently into an active conformation, thereby improving potency. As shown by the superimposition of 5a and 6a (Figure 2(b)), the rigid tetracyclic core of 6a positions the carbonyl oxygen atom and the pyridine ring nearly exactly where they must be to maintain the active conformation of 5a. With this information in hand, we proceeded to synthesis 6a to test our hypothesis that conformation restriction improves potency.
Design strategy of novel tetracyclic chitinase inhibitors. (a) Predicted binding mode of compound 5a with OfChi-h; (b) superimposition of compound 5a and compound 6a; (c) novel conformation restriction strategy by ring closure.
Chemistry
The target compounds 6a–6l were synthesised following the synthetic route depicted in Scheme 1. The starting material, 2-hydroxy-9-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (compound 1), was prepared via a condensation reaction between 2-amino-3-picoline and diethyl malonate at 110 °C. Treatment of compound 1 with phosphorus oxychloride (POCl_3_) in DMF afforded 2-chloro-9-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carbaldehyde (compound 2), which was then reacted with excess aqueous ammonia in ethanol at 70 °C to give the key intermediate 2-amino-9-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carbaldehyde (compound 3). The cyanoacetamide derivatives 4a–4l were synthesised by the condensation of methyl cyanoacetate with the corresponding amines under neat conditions at room temperature. Subsequent condensation of intermediate 3 with cyanoacetamides 4a–4l in the presence of sodium hydroxide furnished compounds 5a–5l via a modified Friedländer reaction. Finally, treatment of 5a–5l with 1,1-dimethoxy-N,N-dimethylmethanamine (DMFDMA) under neat conditions at 110 °C yielded the desired tetracyclic target compounds 6a–6l. All the target compounds were confirmed by NMR spectroscopy and HRMS.
Synthesis of target compounds 6a-l. Reagents and conditions: (a). neat, 110 °C; (b). POCl3, DMF, 0 °C-80 °C; (c). aqueous ammonia, EtOH, 70 °C; (d). methyl cyanoacetate, r.t. (e). NaOH/EtOH, 70 °C; (f). DMFDMA, neat, 110 °C.
Structure-activity relationship analysis
Originally, we sought to condense 5a with DMFDMA to afford 6a. As expected, 6a was obtained and tested against OfChi-h. To our delight, 6a showed a significant increase in inhibitory activity against OfChi-h, with a Ki value of 0.058 μM (Table 1 and Figure 3) compared to that of 5a11 (an approximately 15-fold improvement). The inspiring result motivated further modifications of 6a. Initially, a methyl group was introduced to the side chain of the pyridine to afford 6b (Figure S1). However, this modification unexpectedly resulted in a sharp decrease in inhibitory activity. Due to unsuccessful condensation reactions when larger substituents were introduced on the pyridine side chain, pyridine was replaced by pyrazine to produce compound 6c, in an effort to facilitate additional interactions with OfChi-h. However, 6c showed similar inhibitory activity to 6b (Figure S2). Compound 6d, bearing a phenyl substituent, exhibited further decreased inhibitory activity, with a K_i_ value of 1.419 μM (Figure S3). Though the introduction of fluorine and chlorine (6i) to the phenyl group failed to improve activity, the incorporation of an amino group (6h) and a methoxyl group (6 g) increased activity related to 6d (Figures S4 and S5). Surprisingly, the installation of a second methoxyl group (6f) and a third methoxyl group (6e) further enhanced the inhibitory activity against OfChi-h. 6e showed comparable activity to 6a, with a K_i_ value of 0.065 μM (Figure S3). Other attempts to substitute the pyridine group (6j-6l) also resulted in decreased activity, indicating that the pyridine group may interact with OfChi-h (Figures S4 and S5). We also tried to fuse 5 m to obtain a tetracyclic scaffold, as it showed the most potent activity in our previous study. However, the reaction is sensitive to the side chain of the amide, and 5 m failed to condensed into the corresponding tetracyclic structure due to the presence of a cyclopropyl group—a similar phenomenon was also observed by Wang31. In addition, the most potent compound 6a also displayed good inhibition activity against SmChiA (a bacterial homology of OfChi-h from Serratia marcescens) and HsChit1 (a chitinase from Homo sapiens), with a K_i_ value of 0.126 μM and 0.295 μM, respectively (Figure 3(b,d)).
Representative Dixon plots for inhibition chitinase SmChi-A by 5a (a), and chitinase SmChi-A (b), OfChi-h (c) and HsChit1 (d) by 6a. The trend lines represent three substrate concentrations.
Binding mode of 6a with SmChiA and OfChi-h. A. Binding mode of 6a with SmChiA revealed by co-crystal complex structure; B. Binding mode of 6a with OfChi-h revealed by molecular docking. 6a is shown in violet sticks, the key residues involved in inhibitor binding are shown in gray sticks, hydrogen bonds are shown as black dashed lines and water molecules are shown as red spheres.
Superposition of 6a (a) and 6e (b) with 2–8-s2 in the binding site of OfChi-h. 6a is shown in violet sticks, 6e was shown in red sticks, 2–8-s2 was shown in light green sticks and the key residues involved in inhibitor binding are shown in gray sticks, hydrogen bonds are shown as black dashed lines.
Elucidation of binding modes to OfChi-h
To investigate the binding mode of fused tetracyclic scaffold and to validate our hypothesis, we tried to obtain the crystal structure of OfChi-h and SmChiA in complex with the inhibitor 6a. However, only co-crystal structure SmChiA-6a was successfully obtained and refined. In this structure, compound 6a is bound at the −3, −2 and −1 subsites of SmChiA. The tetracyclic skeleton forms strong π-π interactions with Trp167, and the 11- and 12-nitrogen atoms of 6a form hydrogen bonds with the side chain of Glu473 through a water molecule. One of the carbonyl oxygen atoms in the tetracyclic backbone also forms a hydrogen bond with T276 through water-mediated network, while another carbonyl oxygen atom forms a direct hydrogen bond with the backbone of Trp275. The pyridine moiety inserts into a pocket at the bottom of the binding cleft, which is formed by Tyr390, Met388, and Trp539. The pyridine ring forms π-π stacking and π-S interactions with Trp539 and Met388, respectively. In addition, the nitrogen atom of the pyridine also forms a hydrogen bond with Glu315 through a water-mediated network. Since the SmChiA catalytic cleft is acidic, the nitrogen atom of pyridine moiety may guide the inhibitor to bind in the substrate-binding site.
As we failed to obtain the crystal structure of OfChi-h in complex with 6a, molecular docking was used to investigate its binding mode. The docking results revealed that 6a binds to at the −3, −2 and −1 subsites of OfChi-h in a manner similar to SmChiA. The 3-pyridine group inserts into a binding cleft formed by Tyr156, Phe184, and Met381. The nitrogen atom forms a hydrogen bond with the key catalytic residue Glu308, as observed with inhibitor 2–8-s2 (Figures 4 and 5(a)). This interaction is likely responsible for the higher inhibitory activity against OfChi-h and explains the unsuccessful attempts to replace the 3-pyridine group. The tetracyclic skeleton of 6a forms strong π-π stacking interaction with Trp160, while a second carbonyl oxygen forms a hydrogen bond with backbone of the Trp268. In contrast, molecular docking showed that 6e binds to OfChi-h in a different manner (Figure 5(b)). The tetracyclic skeleton of 6e is sandwiched between Trp268 and Trp389, forming strong π-π stacking interactions similar to that of 2–8-s2. The trimethoxybenzene group inserts into −2 subsites of OfChi-h, because it is too bulky to fit into the binding cleft formed by Tyr156, Phe184, and Met381.
Conclusion
In summary, this study demonstrates the successful development of conformationally restricted dipyridopyrimidine-3-carboxamide derivatives as potent inhibitors targeting OfChi-h. By employing a conformational restriction strategy, we discovered novel OfChi-h inhibitors with significantly improved inhibitory activity. The lead compound 6a exhibited exceptional potency (K_i_ = 58 nM) against OfChi-h, establishing its potential as a sustainable pesticide candidate. Notably, 6a also demonstrated promising activity against human chitinase HsChit1 (K_i_ = 295 nM), suggesting that this scaffold could also be repurposed to target human chitinases implicated in inflammatory diseases10^,^32–34. Structural investigations through molecular docking and high-resolution co-crystallization with SmChiA revealed that 6a binds to OfChi-h via a novel mechanism, occupying subsites −3 to −1 of the substrate-binding cleft. This binding mode not only validates the design rationale but also highlights the advantage of rigidifying the inhibitor scaffold to enhance target engagement. The fused tetracyclic skeleton introduced here serves as a robust structural template for designing selective chitinase inhibitors. Overall, this work underscores the broader utility of conformational restriction as a strategic tool in chitinase inhibitor discovery, bridging rational design with mechanistic validation, and paves the way for broader adoption of conformationally constrained scaffolds in chitinase inhibitors design.
Experimental section
Instruments and chemicals
Unless otherwise noted, standard and analytical graded reagents and solvents were obtained from Bide Pharmatech Ltd (Shanghai, China) and used without further purification. Ethanol, dichloromethane, methanol, and dimethyl sulfoxide were purchased from Shanghai Titan Scientific Co., Ltd. (Shanghai, China), TLC plates (Silica gel 60 F254) and chromatography silica gel (200–300 mesh) were supplied by Qingdao Haiyang Chemical (Qingdao, China), N,N-Dimethylformamide dimethyl acetal (DMFDMA), were sourced from Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). All reactions were under ambient air. Melting points (M.p.) were recorded on Büchi B540 apparatus (Büchi Labortechnik, Flawil, Switzerland) and are uncorrected. High-resolution electron mass spectra (HRMS) were performed on Waters, Xevo G2 TOF spectrometer (MA, USA).^1^H NMR,^19^F NMR and ^13^C NMR spectra were recorded on Bruker AM-400 and Bruker ASCEND 600 MHz (^1^H at 400 MHz, ^13^C at 100 MHz or 150 MHz, ^19^F at 376 MHz) spectrometers with DMSO-d6 or CDCl_3_ as solvents and TMS as internal standard. Chemical shifts are reported in δ (parts per million). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartette, m = multiplet, coupling constant (Hz) and integration.
General synthetic procedure of target compounds 6a-6l
Detailed synthesis procedures of compounds 5a-5l were described in our previous report11. Subsequently, to a round-bottom flask were added compound 5 (0.5 mmol, 1.0 equiv) and 1,1-dimethoxy-N,N-dimethylmethanamine (DMFDMA, 2.0 ml, approx. 15 mmol, ∼30 equiv). The mixture was stirred under neat conditions at 110 °C for 10–60 min (monitored by TLC). After completion, the reaction mixture was cooled to room temperature, and ethanol (2 ml) was added. The resulting solid was collected by filtration, washed with cold ethanol (2 × 1 ml), and dried under vacuum. The crude product was further purified by column chromatography on silica gel using DCM/MeOH (30:1) as eluent to afford the final compounds 6a–6l as yellow solids.
Analysis data of compounds 6a-6l
6a, Yellow solid, Yield: 67.3%, M.p.: 352.1–352.7 °C; ^1^H NMR (400 MHz, DMSO-d6) δ 9.56 (t, J = 5.8 Hz, 1H), 8.93 (s, 1H), 8.74 (d, J = 7.1 Hz, 1H), 8.64 (s, 1H), 8.53 (d, J = 3.8 Hz, 1H), 7.88 (d, J = 7.9 Hz, 1H), 7.78 (d, J = 6.7 Hz, 1H), 7.48 (dd, J = 7.8, 4.9 Hz, 1H), 7.08 (t, J = 7.0 Hz, 1H), 4.52 (d, J = 5.7 Hz, 2H), 2.47 (s, 3H). ^13^C NMR (100 MHz, DMSO) δ 174.45, 166.09, 160.98, 157.66, 150.70, 147.89, 147.19, 143.29, 138.33, 136.58, 136.42, 135.25, 133.18, 125.47, 123.96, 113.42, 110.48, 100.85, 40.33, 17.82. HRMS-ESI (m/z): calcd for C_20_H_15_N_6_O_2_ [M + H]^+^, 371.1256; found, 371.1257.
6b, Yellow solid, Yield: 41.7%, M.p.: 302.5–304.7 °C; ^1^H NMR (400 MHz, Chloroform-d) δ 9.79 (s, 1H), 8.82 (d, J = 7.1 Hz, 1H), 8.74 (s, 1H), 8.63 (d, J = 4.0 Hz, 1H), 8.35 (s, 1H), 7.74 (d, J = 8.1 Hz, 1H), 7.63 (d, J = 6.7 Hz, 1H), 7.37 (dd, J = 7.9, 4.9 Hz, 1H), 6.97 (t, J = 7.0 Hz, 1H), 6.31 (q, J = 7.1 Hz, 1H), 2.70 (s, 3H), 1.95 (d, J = 7.2 Hz, 3H). ^13^C NMR (150 MHz, CDCl_3_) δ 161.48, 160.31, 159.65, 159.35, 151.84, 150.23, 150.01, 148.83, 142.07, 136.21, 135.60, 134.81, 134.45, 125.25, 123.84, 114.69, 113.58, 110.96, 50.79, 29.65, 18.84. HRMS-ESI (m/z): calcd for C_21_H_17_N_6_O_2_ [M + H]^+^, 385.1413; found, 385.1412.
6c, Yellow solid, Yield: 53.4%. M.p.: 306.1–307.0 °C; ^1^H NMR (400 MHz, DMSO-d6) δ 9.25 (s, 1H), 8.87 (d, J = 1.6 Hz, 2H), 8.71 (m, 1H), 8.62 − 8.56 (m, 2H), 7.82 (m, 1H), 7.11 (t, J = 7.0 Hz, 1H), 5.44 (s, 2H), 2.53 (s, 3H).^13^C NMR (100 MHz, DMSO) δ = 161.62, 160.10, 159.17, 158.87, 154.60, 151.13, 150.90, 144.12, 144.01, 143.84, 139.36, 136.86, 133.75, 125.34, 114.14, 113.72, 110.52, 48.35, 18.05. HRMS-ESI (m/z): calcd forC_19_H_14_N_7_O_2_ [M + H]^+^, 372.1209; found, 372.1210;
6d, Yellow solid, Yield: 72.4%, M.p.: 303.8–304.5 °C; ^1^H NMR (400 MHz, Chloroform-d) δ 9.78 (s, 1H), 8.82 (d, J = 7.2 Hz, 1H), 8.45 (s, 1H), 7.62 (d, J = 6.7 Hz, 1H), 7.47 − 7.30 (m, 5H), 6.96 (t, J = 7.0 Hz, 1H), 5.22 (s, 2H), 2.70 (s, 3H). ^13^C NMR (100 MHz, CDCl_3_) δ 160.69, 159.44, 152.56, 141.97, 139.56, 136.15, 135.64, 134.93, 129.90, 129.26, 129.12, 128.76, 128.38, 125.30, 115.18, 113.57, 110.91, 49.85, 29.34. HRMS-ESI (m/z): calcd for C_21_H_16_N_5_O_2_ [M + H]^+^, 370.1304; found, 370.1305;
6e, Yellow solid, Yield: 54.8%, M.p.: 329.8–331.1 °C; ^1^H NMR (400 MHz, Chloroform-d) δ 9.79 (s, 1H), 8.82 (d, J = 7.0 Hz, 1H), 8.45 (s, 1H), 7.62 (d, J = 6.7 Hz, 1H), 6.96 (t, J = 7.0 Hz, 1H), 6.62 (s, 2H), 5.13 (s, 2H), 3.86 (s, 6H), 3.83 (s, 3H).^13^C NMR (150 MHz, CDCl_3_) δ 161.97, 160.62, 159.54, 159.36, 153.76, 152.37, 151.81, 141.96, 138.30, 136.22, 135.56, 130.35, 125.24, 115.04, 113.59, 110.86, 105.56, 60.83, 56.21, 50.04, 18.82. HRMS-ESI (m/z): calcd for C_24_H_22_N_5_O_5_[M + H]^+^, 460.1621; found, 460.1620.
6f, Yellow solid, Yield: 59.1%, M.p.: 303.9–305.1 °C; ^1^H NMR (400 MHz, Chloroform-d) δ 9.78 (s, 1H), 8.81 (d, J = 7.1 Hz, 1H), 8.44 (s, 1H), 7.01 − 6.91 (m, 3H), 6.86 (d, J = 8.1 Hz, 1H), 5.14 (s, 2H), 3.88 (s, 3H), 3.88 (s, 3H), 2.69 (s, 3H). ^13^C NMR (100 MHz, CDCl_3_) δ 162.12, 160.73, 159.59, 159.44, 152.47, 151.83, 149.56, 149.49, 141.91, 136.12, 135.64, 127.34, 125.28, 121.14, 115.17, 113.55, 111.63, 111.44, 110.88, 56.02, 55.98, 49.76, 18.85. HRMS-ESI (m/z): calcd for C_23_H_20_N_5_O_4_[M + H]^+^, 430.1515; found, 430.1514.
6g, Yellow solid, Yield: 68.3%, M.p.: 311.8–314.6 °C; ^1^H NMR (400 MHz, Chloroform-d) δ 9.77 (s, 1H), 8.81 (d, J = 7.1 Hz, 1H), 8.44 (s, 1H), 7.61 (d, J = 6.6 Hz, 1H), 7.36 (d, J = 8.6 Hz, 2H), 6.92 (m,6.87–6.98 3H), 5.15 (s, 2H), 3.80 (s, 3H), 2.69 (s, 3H). ^13^C NMR (100 MHz, CDCl_3_) δ 173.25, 160.43, 157.25, 154.87, 153.95, 149.83, 148.66, 147.36, 135.67, 135.01, 134.18, 133.97, 125.34, 123.44, 113.80, 100.67, 93.58, 38.62, 29.42, 17.89. HRMS-EI (m/z): calcd for C_22_H_17_N_5_O_3_[M^] +^, 399.1331; found, 399.1335.
6h, Yellow solid, Yield: 42.1%. M.p.: 324.6–326.3 °C; ^1^H NMR (400 MHz, Chloroform-d) δ 9.78 (s, 1H), 8.82 (d, J = 6.8 Hz, 1H), 8.42 (s, 1H), 7.62 (d, J = 6.8 Hz, 1H), 7.19 − 7.09 (m, 2H), 6.96 (t, J = 7.0 Hz, 1H), 6.76 (d, J = 7.6 Hz, 1H), 6.71 − 6.59 (m, 3H), 5.11 (s, 2H), 2.70 (s, 3H). ^13^C NMR (150 MHz, CDCl_3_) δ 160.64, 159.52, 159.38, 152.61, 151.76, 147.17, 141.89, 136.07, 136.01, 135.56, 130.15, 129.83, 125.23, 118.19, 115.25, 115.13, 114.50, 113.49, 110.81, 49.64, 29.65. HRMS-ESI (m/z): calcd for C_21_H_17_N_6_O_2_[M + H]^+^, 385.1413; found, 385.1412.
6i, Yellow solid, Yield: 46.2%. M.p.: 353.5–354.5 °C; ^1^H NMR (400 MHz, Chloroform-d) δ 9.77 (s, 1H), 8.82 (d, J = 7.2 Hz, 1H), 8.52 (s, 1H), 7.63 (d, J = 6.7 Hz, 1H), 7.31 − 7.27 (m, 2H), 7.20 − 7.13 (m, 1H), 6.97 (t, J = 7.0 Hz, 1H), 5.34 (s, 2H), 2.70 (s, 3H). ^19^F NMR (376 MHz, DMSO) δ −115.22. ^13^C NMR (150 MHz, CDCl_3_) δ 161.93, 160.63, 159.62, 159.33, 158.47 (d, J = 250.0 Hz), 152.50, 151.84, 141.93, 136.23, 135.61, 134.51, 129.85 (d, J = 5.2 Hz), 128.13 (d, J = 8.0 Hz), 125.94 (d, J = 3.6 Hz),125.24, 116.86 (d, J = 21.3 Hz), 114.95, 113.60, 110.89, 47.21, 29.65. HRMS-ESI (m/z): calcd for C_21_H_14_ ^35^ClFN_5_O_2_ [M + H]^+^, 422.0820; found, 422.0821; calcd for C_21_H_14_ ^37^ClFN_5_O_2_ [M + H]^+^, 424.0791; found, 424.0795.
6j, Yellow solid, Yield: 43.4%. M.p.: 323.4–324.6 °C; ^1^H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H), 9.38 (s, 1H), 8.80 − 8.71 (m, 1H), 8.40 (s, 1H), 7.84 (dt, J = 6.7, 1.4 Hz, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.35 (dt, J = 8.1, 1.1 Hz, 1H), 7.18 (d, J = 2.4 Hz, 1H), 7.13 (t, J = 7.0 Hz, 1H), 7.07 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.95 (ddd, J = 7.9, 7.0, 1.0 Hz, 1H), 4.27 (t, J = 7.3 Hz, 2H), 3.17 (t, J = 7.3 Hz, 2H), 2.54 (s, 3H). ^13^C NMR (150 MHz, DMSO) δ 166.56, 163.44, 160.71, 160.24, 157.97, 153.85, 150.96, 146.18, 139.79, 136.64, 130.06, 129.37, 127.37, 123.88, 122.21, 121.90, 121.49, 118.83, 118.54, 110.40, 110.36, 53.14, 29.38, 18.24. HRMS-ESI (m/z): calcd for C_24_H_19_N_6_O_2_[M + H]^+^, 423.1569; found, 423.1571.
6k, Yellow solid, Yield: 33.4%. M.p.: 297.3–298.5 °C; ^1^H NMR (400 MHz, Chloroform-d) δ 9.76 (s, 1H), 8.81 (d, J = 6.9 Hz, 1H), 8.50 (s, 1H), 7.62 (d, J = 6.7 Hz, 1H), 7.47 − 7.37 (m,1H), 6.96 (t, J = 7.0 Hz, 1H), 6.53 (d, J = 3.1 Hz, 1H), 6.42 − 6.33 (m, 1H), 5.21 (s, 2H), 2.70 (s, 3H). ^13^C NMR (150 MHz, CDCl_3_) δ 161.99, 160.21, 159.51, 159.34, 152.26, 151.79, 147.51, 143.56, 141.86, 136.15, 135.56, 125.23, 115.04, 113.54, 110.87, 110.80, 110.59, 42.13, 18.80. HRMS-ESI (m/z): calcd for C_19_H_14_N_5_O_3_[M + H]^+^, 360.1097; found, 360.1098.
6l, Yellow solid, Yield: 34.2%. M.p.: 200.6.-201.3 °C; ^1^H NMR (400 MHz, Chloroform-d) δ 9.78 (s, 1H), 8.83 (d, J = 7.3 Hz, 2H), 8.67 (s, 1H), 7.64 (d, J = 6.8 Hz, 2H), 6.97 (t, J = 6.8 Hz, 2H), 4.84 (d, J = 2.4 Hz, 3H), 2.71 (s, 3H), 2.57 (s, 1H). ^13^C NMR (150 MHz, CDCl_3_) δ 161.94, 159.93, 159.63, 159.35, 151.86, 151.25, 141.89, 136.25, 135.61, 129.87, 125.26, 114.64, 113.62, 76.09, 75.55, 35.38, 18.80. HRMS-ESI (m/z): calcd for C_17_H_12_N_5_O_2_ [M + H]^+^, 318.0991; found, 318.0995.
Full ^1^H, ^13^C, ^19^F NMR and HRMS raw spectra of compound 6a-6l are included in (Supplementary Material Figures S7–S43).
Preparation and purification of enzyme
SmChiA was expressed in Escherichia coli BL21(DE3), HsChit1 and OfChi-h were overexpressed in Pichia pastoris GS115. The Escherichia coli BL21(DE3) strain was purchased from TaKaRa Biotech (Dalian, 6dChina) while the Pichia pastoris GS115 strain was obtained from Invitrogen (Thermo Fisher Scientific, CA, USA), both of which were cultured and handled according to the protocols provided by suppliers. All chitinases were purified using immobilised metal ion affinity chromatography as previously described35. Briefly, solid ammonium sulphate was slowly added to the culture supernatant to achieve 75% saturation. After incubation at 4 °C for 24 h, the precipitated proteins were collected by centrifugation at 12,000 × g for 30 min at 4 °C. The pellet was resuspended thoroughly in buffer A (20 mM sodium phosphate, 0.5 M sodium chloride, pH 7.4), and the solution was centrifuged again at 12,000 × g for 15 min at 4 °C to remove insoluble material. The clarified supernatant was loaded onto a 5 ml HisTrap^™^ Crude affinity column (GE Healthcare) that had been pre-equilibrated with buffer A. The column was subsequently washed with buffer A containing 75 mM imidazole to remove non-specifically bound proteins. Recombinant chitinases were eluted using buffer A containing 250 mM imidazole. The expressed proteins were quantified using a BCA protein assay kit (TaKaRa Biotech, China). And their purities were determined by SDS-PAGE and found to be >95% in all cases.
Chitinase inhibition assay and Ki determination
Chitinase activities for SmChiA, HsChit1 and OfChi-h were determined using 4-methylumbelliferyl-N, N′-diacetyl-β-D-chitobioside (MU-β-(GlcNAc)2) (purchased from Sigma) as a substrate. The final reaction mixture volume used for inhibitor screening was 100 μL, consisting of 10 nM enzyme, 4 μM MU-β-(GlcNAc)2, 10 μM inhibitor, and 2% DMSO in a 20 mM sodium phosphate buffer (pH 6.0). The reaction in the absence of inhibitor was used as a positive control. After incubation at 30 °C for 20 min, 0.5 M sodium carbonate was added to the reaction mixture, and the fluorescence produced by the released 4-MU was quantified with a Varioskan Flash microplate reader (Thermo Fisher Scientific) using excitation and emission wavelengths of 360 and 450 nm, respectively. Experiments were performed in triplicate unless specified otherwise. For K_i_ value determination, the inhibitor concentration was varied in the above reaction, and different concentrations of substrate (2–8 μM) were used. The amount of released 4-MU was quantified as above, and the K_i_ value and the mode of inhibition were determined using the Dixon plot.
Molecular docking
Protein structure for molecular docking was prepared using the protein preparation wizard in Maestro 10.2 (2015–2 release). The crystal structure of OfChi-h in complex with inhibitor 2–8-s2 (PDB: 6jmn) was used in molecular docking study. Bond orders were assigned, hydrogens were added and waters beyond 5 Å from inhibitor 2–8-s2 were deleted. The protein structure was the refined using PROPKA and minimised OPLS336 force field. The docking grid were generated by Receptor Grid Generation tool integrated in Glide.
The grid box dimensions were determined based on the coordinates of the bound 2–8-s2. A 10 Å10 Å10 Å inner grid box and a 20 Å20 Å20 Å outer grid box was used without other constraints. The 2D structures of 6a-6l were constructed by Chemdraw 17.1 and were imported to Maestro 10.2. Initial ligand conformations are minimised and sampled by MacroModel, which were then prepared LigPrep tool. Hydrogen atoms were added while the ionisation and tautomeric states of ligands at a target pH of 7.0 ± 2.0 were generated using Epik. The 3D geometry of each structure was optimised by OPLS-3 force field. Glide in extra precision (XP) mode was used in docking. Molecular mechanics-generalized Born surface area (MM-GBSA) method in Prime was used for rescoring the docked pose of ligand. For each compound, the pose with the lowest score was retained.
Cocrystallization and data collection
Protein-inhibitor complex crystal was prepared by co-crystallization methods. Briefly, the crystal of SmChiA-6a crystal complex was obtained in a solution consisting of 5 mM 6a, 5% (v/v) DMSO, 0.75 M sodium citrate (pH 7.2) and 20% (v/v) methanol by the vapour method37. The crystals were soaked for 10 s in a reservoir solution containing 20% (v/v) glycerol as a cryoprotection reagent, and subsequently flash-cooled in liquid nitrogen. X-ray diffraction data of the complexes were collected on the BL-19U1 at Shanghai Synchrotron Radiation Facility in China. The diffraction data of the complexes were processed using the HKL-3000 package.
Structure determination and refinement
The structures of SmChiA-6a was solved by molecular replacement with Phaser38 using the structure of free SmChiA as a model (PDB entry 2WLZ). The PHENIX suite of programs39 was used for structure refinement. Coot was used for manually building and extending the molecular models40. The stereo chemical quality of the models was checked by PROCHECK41. The coordinates of SmChiA-6a were deposited in the Protein Data Bank as entry 7FD6. The structural figures were generated using the PyMOL program. The statistics for the diffraction data and the structure refinement are summarised in Table 2.
Supplementary Material
Supplementary Material anonymous.docx
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chen W, Yang Q. Development of novel pesticides targeting insect chitinases: a minireview and perspective. J Agric Food Chem. 2020;68(16):4559–4565.32239934 10.1021/acs.jafc.0c 00888 · doi ↗ · pubmed ↗
- 2Zhu B, Lou M-M, Xie G-L, Zhang G-Q, Zhou X-P, Li B, Jin G-L. Horizontal gene transfer in silkworm, bombyx mori. BMC Genom. 2011;12(1):248.10.1186/1471-2164-12-248PMC 311650721595916 · doi ↗ · pubmed ↗
- 3Liu T, Chen L, Zhou Y, Jiang X, Duan Y, Yang Q. Structure, catalysis, and inhibition of Of Chi-h, the lepidoptera-exclusive insect chitinase. J Biol Chem. 2017;292(6):2080–2088.28053084 10.1074/jbc.M 116.755330 PMC 5313083 · doi ↗ · pubmed ↗
- 4Lu Q, Xie H, Qu M, Liu T, Yang Q. Group h chitinase: a molecular target for the development of lepidopteran-specific insecticides. J Agric Food Chem. 2023;71(15):5944–5952.10.1021/acs.jafc.2c 0884537038745 · doi ↗ · pubmed ↗
- 5Chen W, Jiang X, Yang Q. Glycoside hydrolase family 18 chitinases: the known and the unknown. Biotechnol Adv. 2020;43:107553.32439576 10.1016/j.biotechadv.2020.107553 · doi ↗ · pubmed ↗
- 6Davies GJ, Wilson KS, Henrissat B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem J. 1997;321 (Pt 2)(Pt 2):557–559.9020895 10.1042/bj 3210557 PMC 1218105 · doi ↗ · pubmed ↗
- 7Chen L, Liu T, Duan Y, Lu X, Yang Q. Microbial secondary metabolite, phlegmacin B 1, as a novel inhibitor of insect chitinolytic enzymes. J Agric Food Chem. 2017;65(19):3851–3857.28457127 10.1021/acs.jafc.7b 01710 · doi ↗ · pubmed ↗
- 8Lu Q, Xu L, Liu L, Zhou Y, Liu T, Song Y, Ju J, Yang Q. Lynamicin B is a potential pesticide by acting as a lepidoptera-exclusive chitinase inhibitor. J Agric Food Chem. 2021;69(47):14086–14091.34797675 10.1021/acs.jafc.1c 05385 · doi ↗ · pubmed ↗