Dnmt3a2 expression during embryonic development is required for phenotypic stability
Peter Jones, Minmin Liu, Guillermo Urrutia, Rachel Shereda, Stacey Thomas, Gangning Liang

TL;DR
The Dnmt3a2 isoform is crucial during embryonic development for maintaining proper DNA methylation, which helps prevent abnormal traits later in life.
Contribution
This study reveals the specific roles of Dnmt3a isoforms in DNA methylation and phenotypic stability during development.
Findings
Dnmt3a2 knockout leads to widespread hypomethylation at enhancers, CTCF sites, and imprinted genes during embryogenesis.
Mice lacking Dnmt3a2 show increased sporadic abnormalities like anophthalmia and male infertility.
Hypomethylation of imprinted genes in sperm may explain infertility in Dnmt3a2-deficient mice.
Abstract
Proper function and switching of regulatory elements are essential for the development of vertebrates and is known to be controlled by DNA methylation. We used isoform-specific knockouts of the de novo methyltransferase Dnmt3a, namely Dnmt3a1 and Dnmt3a2, to probe their roles in regulatory element methylation during embryogenesis and postnatal development. Mouse embryos lacking Dnmt3a1 showed minimal loss of methylation, suggesting limited involvement in embryonic development. However, they were smaller than their littermates and died about 4 weeks after birth with considerable postnatal demethylation as previously reported. In contrast, embryos lacking Dnmt3a2 showed widespread hypomethylation particularly at enhancers, CTCF sites and imprinted genes. These methylation deficits were largely repaired after birth, presumably by Dnmt3a1. The mice lacking Dnmt3a2 were viable; however, they…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
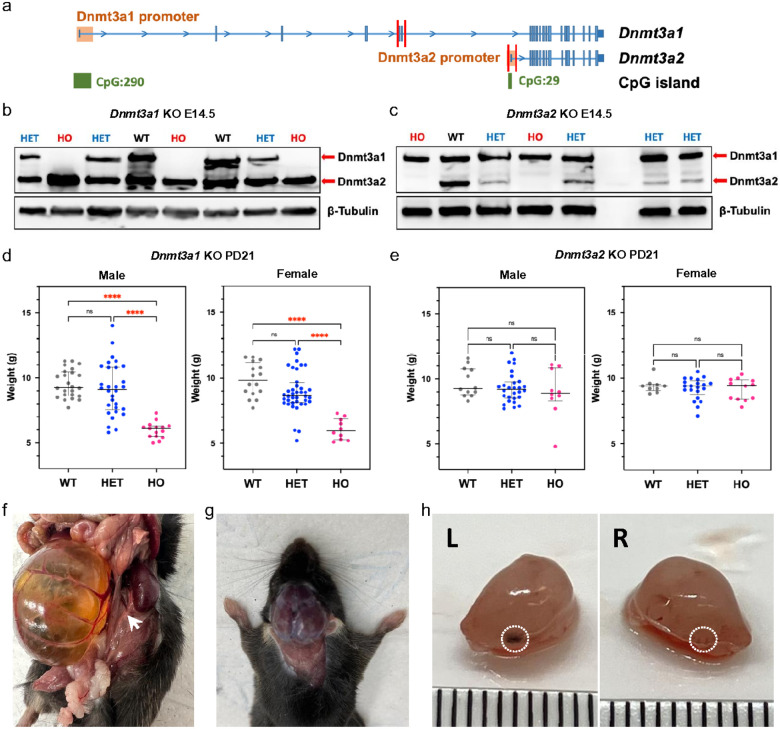 Figure 1
Figure 1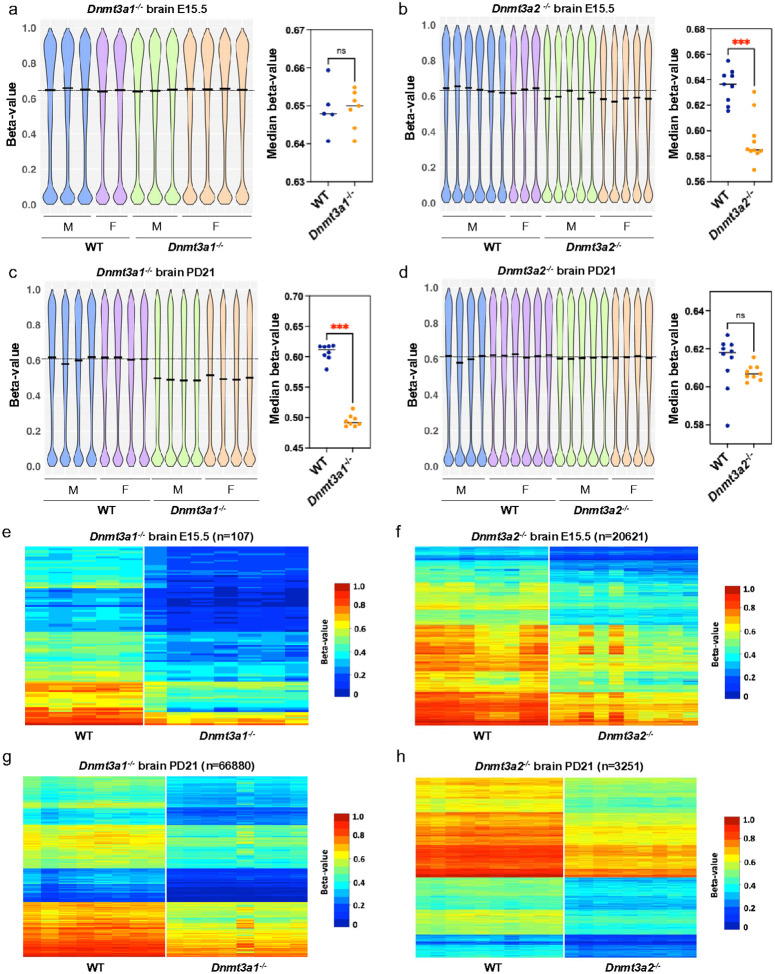 Figure 2
Figure 2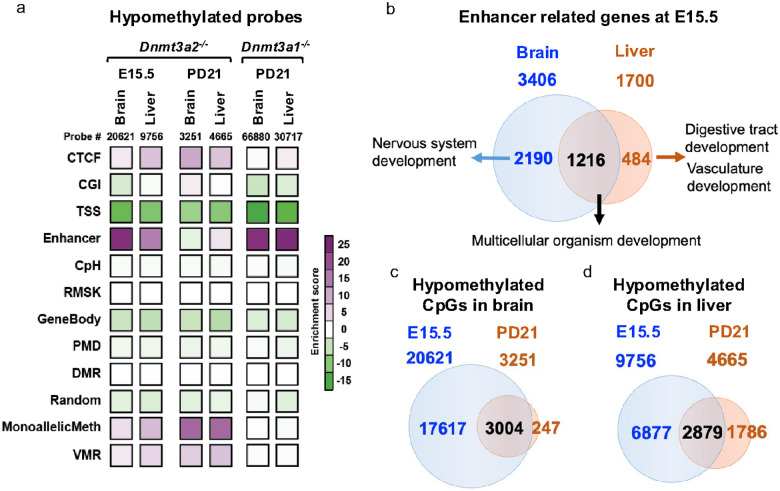 Figure 3
Figure 3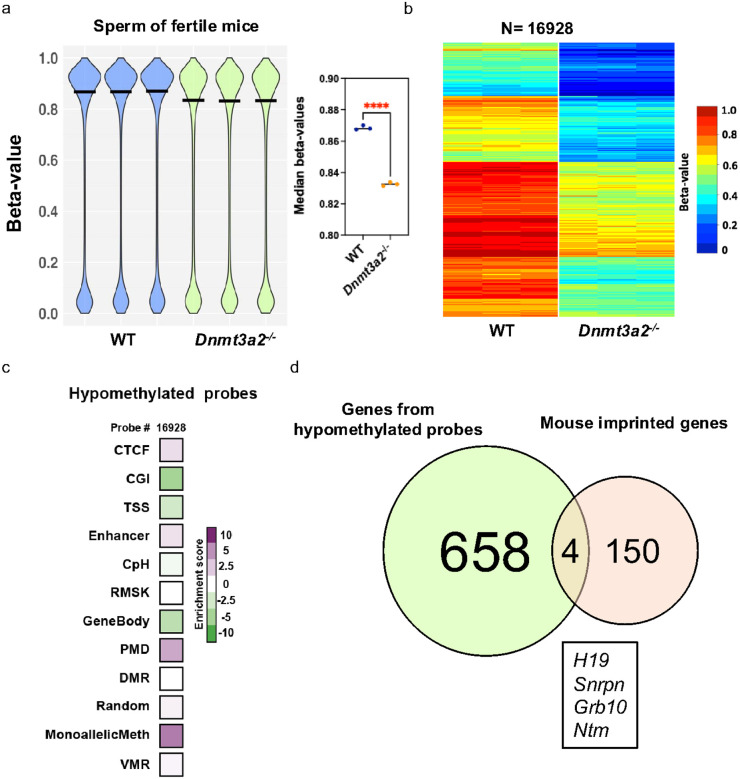 Figure 4
Figure 4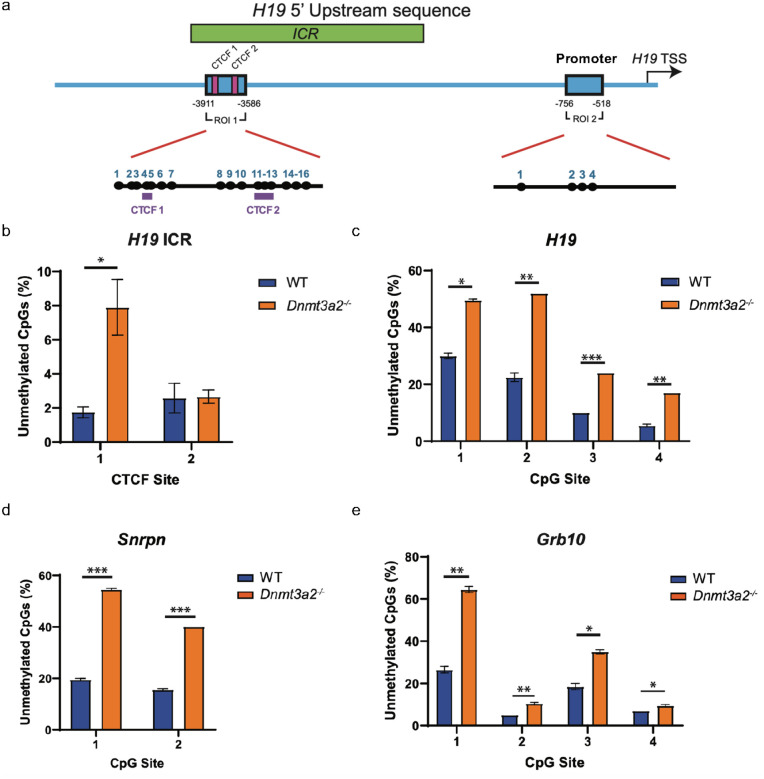 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEpigenetics and DNA Methylation · Genetic Syndromes and Imprinting · Prenatal Screening and Diagnostics
Introduction
DNA methylation is an epigenetic modification that plays a critical role in regulation of gene expression by modulating the activity of key regulatory elements such as promoters, enhancers, CCCTC-binding factor (CTCF) binding sites and Imprinting control regions (ICRs)^1^. These regulatory elements mediate spatial and temporal control of gene expression, ensuring precise transcriptional activation during development^2,3^. The control of gene expression is fundamental to proper formation and function of tissues and organs, as it dictates tissue-specific gene expression patterns^4,5^. However, there is still limited in vivo evidence linking impaired DNA methylation machinery to regulatory element dysfunction particularly for enhancer and imprinted genes, which could ultimately lead to developmental abnormalities.
DNA methylation involves the addition of a methyl group to the 5′ position of cytosine residues within CpG dinucleotides, and is mainly established by the de novo DNA methyltransferases, including Dnmt3a and Dnmt3b, during development in mammals^6^. Mutations in DNMT3A have been implicated in a spectrum of human diseases, particularly hematological malignancies^7–11^ and developmental disorders, including Tatton-Brown-Rahman syndrome (TBRS)^12^ and microcephalic dwarfism^13^. These findings underscore the important role of DNMT3A in normal development and its contribution to disease when dysregulated.
Existing total and tissue-specific Dnmt3a KO mouse models have revealed diverse and context-dependent phenotypes, highlighting the importance of Dnmt3a in various biological processes. For example, homozygous Dnmt3a KO mice (Dnmt3a^−/−^) display severe developmental defects, including growth retardation, premature death, and impaired germ cell development^6^. In contrast, heterozygous Dnmt3a KO mice (Dnmt3a^+/−^) exhibit postnatal phenotypes such as obesity, increased bone length, and behavioral abnormalities, which partially recapitulate human TBRS caused by DNMT3A mutations^14,15^. Tissue-specific KO models further demonstrate that Dnmt3a is essential for germ cell development^16^, hematopoietic stem cell (HSC) differentiation^17^, neurogenesis^18,19^, and suppression of tumorigenesis in a conditional mouse lung tumor model^20^. However, most studies investigating Dnmt3a function using mouse models have not distinguished between its two major isoforms, Dnmt3a1 and Dnmt3a2, which are generated through alternative promoter usage.
Dnmt3a1 features a 219-amino-acid-long N-terminal region, which is missing in Dnmt3a2, while the rest of the protein sequences are the same for both isoforms. The two isoforms exhibit distinct expression patterns, regulatory mechanisms, and potentially unique biological functions^21^. For example, while Dnmt3a1 is ubiquitously expressed and thought to be the predominant isoform in most somatic tissues, Dnmt3a2 is highly expressed in embryonic stem cells (ESCs) and during early development, suggesting it may have specialized roles in maintaining pluripotency or regulating rapid developmental transitions^21^. A recent study has reported isoform-specific mouse models and found that Dnmt3a1, but not Dnmt3a2, is essential for postnatal survival^22^. This highlights the role of Dnmt3a1 in the nervous system regulating bivalent neurodevelopmental genes through interactions of its unique N-terminus with monoubiquitinated histone H2AK119^22^. In addition, Dnmt3a2 has been shown to be essential for maintaining genomic imprinting and epigenomic integrity in mouse embryonic cells^23–25^. These isoform-specific studies have begun to enhance our understanding of their distinct roles in development and disease. Despite their distinct expression profiles and potential functional differences, the individual contributions of these isoforms to methylate different regulatory elements during development remain poorly understood.
To dissect the specific roles of Dnmt3a1 and Dnmt3a2 in modulating the methylation of regulatory elements, we generated isoform-specific knockout models to investigate how each isoform contributes to the establishment and maintenance of methylation patterns during both embryonic and postnatal development. This approach provides an opportunity to unravel the different functions of these isoforms in modulating methylation of regulatory elements leading to eventually distinct developmental phenotypes.
Results
Dnmt3a2 knockout mice exhibit stochastic abnormalities during embryonic and postnatal development
To examine the function of the two different Dnmt3a isoforms during development, we generated isoform-specific Dnmt3a knock-out mouse models. We used CRISPR-based genome editing to target exons 5 and 6 (744 bp) in mouse zygotes to delete Dnmt3a1 specifically (Fig. 1a), resulting in frame shift and premature termination during translation (Extended Data Fig. 1). To delete Dnmt3a2, we targeted the promoter and exon1 of Dnmt3a2 (941 bp), which are located within the intron 6 of the Dnmt3a1 locus (Fig. 1a). Dnmt3a1 or Dnmt3a2 proteins were completely depleted in the respective E14.5 homozygous knockout embryos without affecting the expression of the other isoform (Fig. 1b, c). As previously reported^22^, the growth of Dnmt3a12^−/−^ mice was delayed, as shown by the reduced body weight compared to wildtype (WT) mice at postnatal day 21 (PD21) (Fig. 1d). All Dnmt3a1^−/−^ pups were runted followed by death between 3 and 4 weeks after birth, showing similar phenotypes as the Dnmt3a-null mice^6,22^. The body weight of both male and female Dnmt3a1^+/−^ mice exhibited a bimodal distribution, with increased variation compared to their WT littermates at PD21 (Fig. 1d), similar to previously reported phenotypes of Dnmt3a^+/−^ mice^26^. The observation that Dnmt3a1 KO mice phenocopied Dnmt3a KO mice suggests that Dnmt3a1 is the predominant isoform and plays a critical role in postnatal development.
Although the male and female Dnmt3a2^−/−^ mice were viable and their body weights showed no differences compared to their WT and Dnmt3a2^+/−^ littermates at PD21 (Fig. 1e), sporadic Dnmt3a2^−/−^ mice exhibited reduced (50%) bodyweight (4.5%, Fig. 1e). We also observed the incidence of sporadic developmental defects which were manifested from 14.5 days during embryonic development to 6 months postnatal (Fig. 1f–h). These include hydronephrosis due to unilateral ureteral agenesis with a frequency of 5 out of 203 (2.5%) Dnmt3a2^−/−^ mice (Fig. 1f and Table 1). The incidence of hydrocephalus was 2 out of 203 (1%) (Fig. 1g and Table 1), characterized by the entire brain being filled with cerebrospinal fluid. True anophthalmia also occurred in 4 out of 203 (2%) Dnmt3a2^−/−^ mice, and 1 out of 560 (0.18%) Dnmt3a2^+/−^ mice, characterized by the complete absence of one or both ocular structures due to failed optic vesicle formation during early embryogenesis (Fig. 1h and Table 1). In addition, out of 70 actively breeding males, 4 were identified as infertile, exhibiting an inability to successfully sire offspring despite confirmed mating activity, corresponding to an infertility rate of 5.7% within the tested cohort (Table 1). Collectively, the cases for the stochastic developmental defects were over 15% in Dnmt3a2^−/−^ mice, which were not found in the WT or Dnmt3a1^+/−^ mice (Table 1), although some developmental defects have previously been reported in the heavily inbred C57BL6 background (https://www.informatics.jax.org/inbred_strains/mouse/docs/C57BL.shtml). The observation that knocking out Dnmt3a2 increases the incidence of developmental defects in a stochastic manner suggests a role of the Dnmt3a2 to ensure phenotypic stability during embryonic development.
Dnmt3a2 participates in de novo methylation during embryonic development
We next investigated the DNA methylation changes that might be associated with the stochastic developmental defects observed in Dnmt3a2^−/−^ mice compared to their WT litter mates, focusing on two critical developmental windows: E15.5, and 21 days postnatal (PD 21). These time points were selected to capture methylation dynamics during peak embryonic DNA methylation establishment (E15.5) and during postnatal tissue maturation (PD 21). To examine tissue-specific DNA methylation patterns, we isolated brain and liver tissues and used the Illumina MM285 Infinium Methylation EPIC array to perform genome-wide methylation analysis. Methylation values for each probe (CpG site) were expressed as beta values (β-value), defined as the ratio of methylated probe intensity to overall probe intensity^27^. We first examined the overall DNA methylation pattern in both male and female Dnmt3a1^−/−^ and Dnmt3a2^−/−^ mice compared to their WT littermates by visualizing the distribution of β-values for all autosomal probes. At E15.5, the median levels of DNA methylation for brain and liver in WT mice are 0.64 and 0.50, respectively, showing tissue-specific methylation patterns (Fig. 2a and Extended Data Fig. 2a). While Dnmt3a1^−/−^ embryos did not exhibit overall methylation changes (Fig. 2a and Extended Data Fig. 2a), a significant reduction (p < 0.001) of the median DNA methylation levels were observed in Dnmt3a2^−/−^ embryos (0.585 and 0.478 for brain and liver, respectively), showing hypomethylation in Dnmt3a2^−/−^ embryos (Fig. 2b and Extended Data Fig. 2b). These data indicate that Dnmt3a2 contributes to establish de novo methylation during embryonic development, while Dnmt3a1 is dispensable, despite being highly expressed at this stage (Fig. 1b). However, at PD21, substantial hypomethylation (p < 0.001) for Dnmt3a1^−/−^ mice was observed (Fig. 2c and Extended Data Fig. 2c), while the overall hypomethylation in Dnmt3a2^−/−^ mice was not significant (p = 0.0653 for both brain and liver) (Fig. 2d and Extended Data Fig. 2d). We also used a liquid chromatography-mass spectrometry (LCMS) method to measure the global 5mC level at PD21 and found a significant reduction (p < 0.0001) of 5mC level in the brains of Dnmt3a1^−/−^ mice but not in the liver (Extended Data Fig. 3), possibly because site specific methylation patterns may not be captured at the global level.
To further quantify the level of hypomethylation, we used 0.1 β-value difference and p < 0.05 as a cutoff to select the probes that showed consistent hypomethylation in our KO tissues compared to WT (Fig. 2e–h). In Dnmt3a1^−/−^ mice, minimal hypomethylation was detected at E15.5 in both brain (0.041%) and liver (0.018%) (Fig. 2e, Extended Data Fig. 2e and Table 2). However, by PD21, the level of hypomethylation escalated dramatically, reaching 26% in brain and 12% in liver, reflecting a profound postnatal decrease in the level of methylation (Fig. 2g, Extended Data Fig. 2g and Table 2). This progressive lack of methylation suggests that Dnmt3a1 is dispensable during early embryonic development for de novo methylation but becomes critical for establishing methylation during postnatal maturation. Tissue-specific differences were pronounced, with brain exhibiting double the hypomethylation of liver, underscoring a heightened reliance on Dnmt3a1 in neural tissue for maintaining postnatal epigenetic stability as previously published^22^.
In contrast, Dnmt3a2^−/−^ mice displayed distinct embryonic-predominant hypomethylation. Applying the same beta-value cutoff (≥ 0.1 reduction vs. wildtype and p < 0.05), significant embryonic hypomethylation was observed at E15.5 in brain (8.1%) and liver (3.8%) (Fig. 2f, Extended Data Fig. 2f and Table 2), consistent with Dnmt3a2’s role in de novo methylation during organogenesis. Postnatally, hypomethylation levels declined to 1.3% in brain and 1.8% in liver by 21 days (Fig. 2h, Extended Data Fig. 2h and Table 2), suggesting partial recovery of DNA methylation by Dnmt3a1 after birth. This temporal reversal implies that Dnmt3a2 is indispensable for establishing methylation patterns during embryogenesis, but its absence is partially mitigated postnatally, likely through compensatory mechanisms. Taken together, these findings suggest that Dnmt3a2 is mainly responsible for establishing de novo methylation during embryonic development (E15.5), while Dnmt3a1 plays the dominant role during the postnatal stage (PD21).
Dnmt3a2 ensures proper de novo methylation especially at enhancers during embryonic development
As we mentioned previously, Dnmt3a2^−/−^ mice exhibit stochastic phenotypes during embryonic and postnatal development. We then further investigated the genomic regions where the hypomethylated probes regulated by Dnmt3a2 are located, using classified genomic elements designed in the MM285 array^27^. We then calculated the enrichment of hypomethylated probes in these genomic elements against their overall distribution in the mouse EPIC arrays. We found that the hypomethylated probes were highly enriched in enhancers, CTCF sites and monoallelic methylation regions in Dnmt3a2^−/−^ brain and liver at E15.5 (Fig. 3a). Interestingly, the enrichment for enhancers disappeared mostly at PD21, but the CTCF sites and imprinted genes (MonoallelicMeth) were still enriched (Fig. 3a). In contrast, Dnmt3a1^−/−^ brain and liver exhibit hypomethylation highly enriched exclusively for enhancers after birth (Fig. 3a). These results suggest that Dnmt3a2 is important for establishing DNA methylation at enhancers and CTCF binding sites during embryonic and/or postnatal development. In contrast, the observed enrichment of hypomethylation at imprinted genes (MonoallelicMeth) may result from defects in de novo methylation establishment in the gametes of the Dnmt3a2^+^/^−^ parental mice. Although Dnmt3a2 is important to establish DNA methylation during embryonic development, its activity on enhancers could be fully compensated by the function of Dnmt3a1 after birth, but Dnmt3a2’s activity on CTCF sites and imprinted genes cannot be compensated by Dnmt3a1 after birth.
As previously established, enhancers are tissue-specific, and our findings further demonstrate that Dnmt3a2 regulates enhancer DNA methylation in a tissue-specific manner. At E15.5, the enhancer related genes identified in brain and liver of the Dnmt3a2^−/−^ mice were partially overlapped (Fig. 3b). We analyzed the gene ontology (GO) of enhancer-related genes using functional clustering of the GO terms and found that the overlapping genes in brain and liver were largely involved in multicellular organism development processes, while the other half of the hypomethylated enhancers in Dnmt3a2^−/−^ embryos are near genes driving tissue specific development (Fig. 3b). Intriguingly, these methylation deficits were largely repaired after birth (Fig. 3c, d), possibly due to the compensatory activity of the remaining DNA methyltransferases, including the Dnmt3a1 isoform. The majority of probes remaining hypomethylated in PD21 Dnmt3a2^−/−^ brain and liver were enriched for CTCF sites and imprinted genes (Fig. 3a). This tissue-specific methylation defect of development-related enhancers during embryonic development in Dnmt3a2^−/−^ mice likely increases the chances of stochastic developmental defects observed in a subset of animals.
Dnmt3a2 loss reduces the methylation of infertility-related imprinted genes in sperm.
Considering that male infertility was the most frequent phenotype in Dnmt3a2^−/−^ mice (Table 1), and that the expression of this isoform remains present in adult mice testes^22^, we hypothesized that the loss of Dntm3a2 might affect the DNA methylation profile in mouse sperm. For the four infertile Dnmt3a2^−/−^ mice, we failed to isolate any motile sperm from the one example we examined. We therefore isolated motile sperm from fertile WT and Dnmt3a2^−/−^ mice and evaluated the DNA methylation patterns for all the autosomal probes using the EPIC array. Using a beta-value cutoff of ≥ 0.2 and p < 0.05, we found that Dnmt3a2^−/−^ sperm showed significant hypomethylation (6.5% of total), (Fig. 4a, b). The hypomethylated probes were enriched in four genomic elements: Monoallelic methylation (5.9%), PMD (4.0%), CTCF (1.6%), and Enhancer (1.4%) (Fig. 4c), indicating that Dnmt3a2 is required for complete DNA methylation of multiple regions in spermatozoa.
Imprinted genes are known to play a critical role in spermatogenesis, and alteration in their methylation can impede normal spermatogenesis^28–30^. Considering the importance of Dnmt3a2’s activity on monoallelic methylation in the process of gene imprinting (Fig. 3a), we further investigated if imprinted genes were hypomethylated in sperm from Dnmt3a2^−/−^ mice. We identified 1498 hypomethylated probes distributed over 658 genes in the monoallelic methylation category (Fig. 4d). We compared the hypomethylated genes against a database of 150 previously described mouse imprinted genes and found 4 hypomethylated imprinted genes in Dnmt3a2^−/−^ sperm: H19, Snrpn, Grb10 and Ntm (Fig. 4d). These data indicates that Dnmt3a2^−/−^ mice display a gene imprinting defect in sperm. Interestingly, DNA methylation defects of H19 and Snrpn have been previously associated with male infertility^30^, suggesting that Dnmt3a2 expression is necessary for proper methylation of imprinted genes associated with sperm development and fertility.
Dnmt3a2 KO-induced methylation loss in sperm DNA follows a stochastic pattern.
We examined the hypomethylated regions of three imprinted genes using targeted amplicon bisulfite sequencing to include adjacent CpG sites not present in the array probes. For the H19 gene, we confirmed that Dnmt3a2^−/−^ sperm showed greater hypomethylation of the CTCF1 (8%) compared to WT (2%), but not the CTCF2 binding site of the ICR (Fig. 5a, 5b and Extended Data Fig. 4). Similarly, the region located at the promoter of H19 showed significant hypomethylation of the 4 CpG sites evaluated (Fig. 5a, 5c and Extended Data Fig. 5). In the Snrpn, and Grb10, each CpG site evaluated showed a different degree of hypomethylation compared to the controls. There was a 2-to-3-fold change increase in the level of hypomethylation for most sites (Fig. 5d, 5d, Extended Data Figs. 6 and 7). Next, we evaluated the DNA methylation patterns of these regions in individual DNA sequences and observed that DNA methylation loss did not follow a specific pattern (Extended Data Fig. 4–7). The number of unmethylated CpG sites per sequence was variable, and they appeared randomly distributed within the sequence. Additionally, our data shows that Dnmt3a2^−/−^ mice exhibit DNA methylation defects to be stochastically distributed within the sperm population, as evidenced by the multiple unique methylation patterns shown in the DNA sequences evaluated (Fig. S4 to S7). Previous reports have confirmed the expression of Dnmt3a2 isoform during the latter stages of sperm maturation^31^. Our data further supports the need for Dnmt3a2 expression during spermatogenesis, suggesting an active role for Dnmt3a2 at completing the methylation of multiple genomic regions in sperm, more importantly in imprinted genes related to male infertility, such as H19.
Discussion
Our findings highlight a developmentally regulated interplay between the Dnmt3a1 and Dnmt3a2 isoforms, Dnmt3a2 plays a central role in securing proper enhancer, CTCF sites and imprinted gene methylation during embryogenesis, while Dnmt3a1 focuses on securing proper enhancer methylation in the postnatal stage of development. The compensation of methylation deficits after birth by Dnmt3a1 suggests a division of labor between the isoforms, with Dnmt3a2 acting as the primary de novo methyltransferase during early development and Dnmt3a1 completing DNA methylation patterns postnatally. The increased incidence of sporadic phenotypes in Dnmt3a2-deficient mice underscores the potential importance of proper enhancer methylation in ensuring robust developmental outcomes. This study provides new insights into the isoform-specific functions of Dnmt3a and emphasizes the need to consider these distinct roles when investigating the mechanisms underlying functional elements, such as enhancers, CTCF sites and ICRs, and developmental disorders.
The distinct functional roles of Dnmt3a isoforms during development cannot be fully explained by their different expression patterns alone. While our data demonstrate equivalent expression levels of Dnmt3a1 and Dnmt3a2 during embryogenesis in WT mice (Fig. 1b, c), their knockout models reveal different impacts on DNA methylation. Specifically, Dnmt3a2^−/−^ embryos exhibit significant hypomethylation at E15.5, whereas Dnmt3a1^−/−^ embryos show negligible methylation defects at this stage (Table 1). This suggests that Dnmt3a2 is the dominant isoform responsible for establishing de novo methylation during early embryonic development, likely through its collaboration with Dnmt3L to target enhancers and CTCF-binding sites, as well as imprinted loci in the parental gametes^16,32,33^. In contrast, our data also demonstrates that Dnmt3a1 becomes indispensable postnatally, particularly at bivalent promoters and enhancers, where its loss provides the explanation of severe developmental abnormalities^22^. Our findings underscore a division of labor between the two Dnmt3a isoforms and highlights the subtle regulation of DNA methylation landscapes necessary for robust developmental outcomes.
The sporadic phenotypes observed in Dnmt3a2^−/−^ mice further underscore the importance of methylation fidelity during embryogenesis. The incomplete penetrance of developmental defects in these mice suggests that Dnmt3a2-mediated methylation at enhancers acts as a buffer against intrinsic developmental heterogeneity—a phenomenon that is stochastic in nature^26,34,35^—thereby promoting robust developmental outcomes and limiting phenotypic variability. This is supported by our methylation analysis of spermatozoa from fertile Dnmt3a2^−/−^mice, which revealed stochastic hypomethylation at ICRs, promoters and enhancers of imprinted genes compared to WT controls (Fig. 5 and Extended Data Fig. 4–7), potentially explaining the observed sporadic cases of male infertility^16,28–30^. While bulk methylation assays in E15.5 brain and liver tissues (using array-based approaches) reflect averaged methylation levels across heterogeneous cell populations (Fig. 2), we can infer that Dnmt3a2 loss results in cell-to-cell variability of DNA methylation at enhancers during embryogenesis. Such stochastic hypomethylation likely disrupts the precision of transcriptional regulation in individual cells, ultimately manifesting as sporadic developmental phenotypes in Dnmt3a2^−/−^mice. Taken together, the loss of Dnmt3a2-mediated epigenetic regulation during embryogenesis appears to destabilize developmental processes, increasing the possibility of subsequent abnormalities.
The sporadic phenotypes observed in Dnmt3a2^−/−^mice, including anophthalmia, hydrocephalus, and hydronephrosis, which reflect profound developmental defects, such as the complete absence of the ureter (hydronephrosis), eye (anophthalmia), and potentially ciliary dysfunction (hydrocephalus), can be attributed to DNA methylation changes caused by the loss of Dnmt3a2 during critical developmental windows, rather than genetic mutations or environmental causes. For example, hydronephrosis in inbred C57BL/6 mice has been linked to autosomal recessive mutations in Aqp2cph, which induce nephrogenic diabetes insipidus and polyuria, leading to obstructive nephropathy without structural deficits in the pyeloureteral peristaltic machinery^36–38^. In such models, hydronephrosis arises postnatally due to functional urinary tract obstruction rather than developmental absence of the ureter. By contrast, the complete agenesis of the ureter in Dnmt3a2^−/−^mice implicates a failure in early developmental patterning, likely stemming from disrupted epigenetic regulation during critical morphogenetic windows. In addition, the incidence of hydrocephalus in Dnmt3a2^−/−^ mice was 1% (2 out of 203), a 34-fold increase compared to the background level of 0.029% in the inbred C57BL/6J strain (https://www.jax.org/news-and-insights/2003/july/hydrocephalus-in-laboratory-mice), strongly implicating Dnmt3a2 loss as a contributing factor. Similarly, the observed eye abnormalities, which may result from defects in lens development, and the severe hydronephrosis phenotype, which cannot be explained by genetic mutations alone, further support the role of epigenetic dysregulation in these defects. The fact that environmental factors like alcohol and other teratogens can exacerbate the rate of developmental defects suggests that epigenetic mechanisms, in addition to genetic factors, play a critical role in shaping developmental outcomes. Future studies mapping methylation dynamics at single-cell resolution in Dnmt3a2^−/−^ embryonic tissues could directly test whether stochastic or uniform hypomethylation at lineage-specific enhancers underlies these phenotypes.
In summary, this study provides new insights into the isoform-specific functions of Dnmt3a and highlights the importance of considering these distinct roles when investigating the epigenetic regulation of development and disease. The observed phenotypes in Dnmt3a2-deficient mice emphasize the potential consequences of enhancer dysregulation and suggest that Dnmt3a2 may serve as a safeguard against developmental instability. Future studies should explore the molecular mechanisms underlying the isoform-specific functions of Dnmt3a and their potential implications for human developmental disorders and diseases linked to epigenetic dysregulation. Overall, this work advances our understanding of the complex roles of Dnmt3a isoforms in shaping the epigenetic landscape and ensuring proper development.
Methods
Animals and husbandry
This research complies with ethical regulations, with protocols approved by the Institutional Animal Care and Use Committee (Van Andel Institute (VAI); protocols 20-06-006 and 23-06-013).
The Dnmt3a1 and Dnmt3a2 KO mice were generated by injecting CRISPR–Cas9/sgRNA mixtures into B6C3F2 zygotes and were performed by the VAI Transgenics Core (RRID:SCR_022914). Briefly, 50 ng/ul HiFi AltR Cas9 (Integrated DNA Technologies), 25 ng/ul crRNA:tracrRNA1 and 25 ng/ul crRNA:tracrRNA2 (Integrated DNA Technologies) mixed in modified Tris-EDTA (TE) buffer (10mM Tris-HCl pH 7.4, 0.1mM EDTA) was injected into B6C3F2 zygote cytoplasm. Sequences of gRNA are listed in Supplementary Table 1. Mosaic mice from injections were then back crossed to WT C57BL/6J mice for at least seven generations to obtain heterozygous mice. The homozygous mice were generated by intercrossing the heterozygotes. Littermate controls were used for all comparisons between WT and mutants. For genotyping, ear punch biopsies were collected and the primers used are listed in Supplementary Table 1. All mice were housed at VAI Vivarium core (RRID:SCR_023211) in individual ventilated cages (Tecniplast, Sealsafe Plus GM500 in DGM Racks) with a 12-h/12-h light/dark cycle at 22 ± 1 °C and 30%–70% humidity, and were fed breeder chow (LabDiet, 5021, 0006540). Mice were checked daily by animal keepers and two to three times per week by expert VAI Vivarium Core Staff for health, well-being and any health concerns. Mice with reported phenotypes were euthanized via CO2 asphyxiation.
Tissue lysate preparation and western blot analysis
To examine Dnmt3a1 and Dnmt3a2 expression, brain tissue from E14.5 embryos were crushed and lysed in 200 μl T-PER^™^ Tissue Protein Extraction Reagent (Thermo Scientific) supplemented with 2% SDS and 1× protease inhibitors (Roche). The extracted proteins were denatured in SDS-loading buffer and analyzed by western blot using DNMT3A (E9P2F) Rabbit mAb (Cell Signaling Technology CST 49768) and b-tublin (D3U1W) Mouse mAb (Cell Signaling Technology CST-86298).
Tissue preparation and DNA extraction
Brain (right cerebrum) and liver from E15.5 embryo and PD21 mice were digested in lysis buffer (10mM Tris-HCl pH 8.0, 10mM EDTA, 1% SDS, and 2 mg/ml Proteinase K) overnight at 55°C, followed by phenol-chloroform 1:1 isolation and 50% isopropyl alcohol precipitation. DNA pellet was then washed twice in 70% ethanol and dissolved in TE buffer (10mM Tris-HCl pH 8.0, 0.1mM EDTA).
DNA isolation from fertile mice sperm
Cauda epididymis from WT and fertile Dnmt3a2^−/−^mice were removed and cleaned before suspending them in M16 buffer (95mM NaCl; 4.8mM KCl; 1.1mM KH2PO4, 1.2mM MgSO4, 0.15% Sodium lactate; 5.5mM Glucose, 2.1mg/ml NaHCO3, 0.36mg/ml Sodium pyruvate, 2.3mM CaCl2, and 4mg/ml BSA). Cauda epididymis was nicked, transferred to a tube, and incubated in M16 buffer for 2 hours in an incubator (37°C, 5% CO2) to allow healthy sperm to swim up. Supernatant containing sperm was removed without disrupting the bottom and sperm spun down by centrifugation (3000 rpm, 6 min). Sperm was digested in lysis buffer (10mM Tris-HCl pH 8.0, 10mM EDTA, 1% SDS, 0.1M DTT and 2 mg/ml Proteinase K) overnight at 55°C, followed by phenol-chloroform 1:1 isolation and 100% ethanol precipitation. DNA pellet was then washed twice in 70% ethanol and dissolved in TE buffer (10mM Tris-HCl pH 8.0, 0.1mM EDTA).
Mouse DNA methylation array
DNA samples (20–500 ng) were bisulfite converted using the Zymo EZ DNA Methylation Kit (Zymo Research) following the manufacturer’s protocol with modifications for the Illumina Infinium methylation assays. DNA methylation was assessed using Illumina Infinium Mouse Methylation BeadChip array (MM285), conducted by the VAI Genomics core (RRID:SCR_022913) following the manufacturer’s specifications. Arrays were scanned on the Illumina iScan platform, and probe-specific calls were made using Illumina GenomeStudio version 2011.1 to generate IDAT files.
DNA methylation analysis
Quality control, preprocessing and normalization process were performed using the SeSAMe pipeline (version 1.24.0) on Bioconductor^39^, and the methylation status of individual CpG site was reported as a b-value, ranging from 0 (unmethylated) to 1 (fully methylated). Probes located on the sex chromosomes were excluded from the analysis to enable comparison between KO and WT tissues independent of sex. Probe enrichment analysis was performed using the SeSAMe knowYourCG module^39^, with annotations based on the knowYourCG tool, and related information available at http://zwdzwd.github.io/InfiniumAnnotation#mouse. The ChromHMM annotation was derived from a mouse consensus by the ENCODE project profiling 66 mouse epigenomes across 12 tissues at daily intervals from embryonic day 11.5 to birth^40^. Gene ontology analysis of probe-enriched genes was performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) functional annotation clustering (v2023q4)^41^. Further data visualization of SeSAMe output was performed using RStudio (2024.12.1.563) in R (version 4.4.2).
Nucleoside Quantitation in DNA digests by LC-MS
Absolute nucleoside quantitation in DNA digests was accomplished using liquid chromatography (LC)-mass spectrometry (MS) in the Van Andel Institute Mass Spectrometry Core (RRID:SCR_024903). An external calibration curve was prepared from a stock mix containing 2’deoxycytidine (dC; 100 μg/mL) (30125, Caymen Chemical), 5-methyl-2’deoxycytidine (5mdC; 4 μg/mL) (16166, Caymen Chemical), and 2’deoxyguanosine (dG; 100 μg/mL) (9002864, Caymen Chemical). The stock mix underwent a 7-step serial dilution of half-log steps for eight total curve points. 35μL of DNA digests and standard curve samples were extracted with 315μL of 80% methanol (v/v) containing [13C515N]2’deoxycytidine (6.3 ng/mL) (D239552, Toronto Research Chemicals), [D3]5-methyl-2’deoxycytidine (12.6ng/mL) (M295902, Toronto Research Chemicals), and [13C5] 2’deoxyguanosine (12.6ng/mL) (D232617, Toronto Research Chemicals) as internal standards.
Samples and standards were analyzed with an Agilent 6470 triple quadrupole mass spectrometer coupled with an Agilent ultra-high performance liquid chromatography 1290 Infinity II. 2 μL of each sample was injected separated using a 21-minute gradient on a Cortecs T3 Column (1.6 μm, 2.1mm × 150mm, 186008500, Waters, Eschborn, Germany) combined with a Cortecs VanGuard cartridge (1.6 μm, 2.1 mm × 5 mm, 186008508, Waters). Mobile phase A consisted of LC/MS grade water (W6, Fisher) with 1mM ammonium acetate (73594, Sigma), and 0.01% ammonium hydroxide (A470, Fisher). Mobile phase B consisted of 99% Acetonitrile (A955–4, Fisher) and 1% LC/MS grade water (W6, Fisher). Column temperature was kept at 30 °C, flow rate was held at 0.3 mL/min, and the chromatography gradient was as follows: 0–6 min held at 0% B, 6–9 min ramp from 0 to 10% B, 9–14 min ramp from 10% to 50% B, 14 to 18.9 min ramp from 50% to 99% B. At 19 min, flow is changed to 0% B at 0.4 ml/min, and held until 20.5 min, then decreased to 0.3 ml/min by 21 min. Mass spectrometer parameters were: Gas flow at 13 L/min at 80°C, sheath gas flow at 11 L/min at 275 °C and the nebulizer was set to 30 psi. Capillary voltage was +2500 and nozzle voltage was +500. Data were acquired using dynamic multiple reaction monitoring (dMRM) including at least two transition per compound. The transition list was developed and optimized using neat analytical standards and parameters are provided in Supplemental Table 2. The dMRM parameters were determined based on running LC/MS grade analytical standards for each target compound. Peak picking and integration was performed using Skyline Software (v 24.1). The ISTD peak area was used to correct for matrix effect differences between the samples and standards. The corrected peak areas were used to generate a linear or quadratic regression for quantitation of the target analytes. The quantified analytes (ng/mL) were then used to calculate 5mdC/dG ratio.
Targeted amplicon bisulfite sequencing and analysis
Bisulfite conversion of DNA was carried out using the EZ DNA Methylation-Gold Kit (Zymo Research, #D5006) according to manufacturer’s recommendations. H19 ICR region was amplified using a nested PCR approach. Briefly, 100ng of bisulfite-converted DNA was amplified with primer set 1 using ZymoTaq Polymerase (Zymo Research, #E2002) under the following amplification conditions: 95°C for 10 minutes, 35 cycles of amplification (95°C for 30 seconds, 54°C for 30 seconds, 72°C for 40 seconds), final extension at 72°C for 7 minutes. Second round of amplification was carried out using template from first reaction with the primer set 2 using same amplification conditions. H19, Snrpn, and Grb10 regions were amplified using MyTaq Red Mix (Meridian Biosciences, BIO-25044) under the following amplification conditions: 95°C for 2 minutes, 40 cycles of amplification (95°C for 30 seconds, 58°C for 30 seconds, 72°C for 60 seconds), final extension at 72°C for 5 minutes. Primers used are listed in supplementary table 1. Bisulfite PCR products were sequenced using the Genewiz Amplicon-EZ sequencing service (Azenta Life Sciences) and aligned to reference sequences using the BISulfite-seq CUI Toolkit^42^. BAM files from sorted sequences were created using SAMtools^43^ and loaded in IGV 2.17.4 for visualization and calculation of CpG dinucleotide methylation percentage.
Statistical analysis
Experimental data were analyzed using Prism 10 software (GraphPad, version 10.4.1) by one-way ANOVA, two-tailed Student’s t-test or indicated in the corresponding figure legends. Statistical significance levels are denoted as follows: *,p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, ns, not significant. Data are presented as median ± 95%CI or mean ± s.e.m. as indicated in the corresponding figure legends. No statistical methods were used to predetermine sample size.
Supplementary Material
Supplementary Files
This is a list of supplementary files associated with this preprint. Click to download.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jones P.A.: Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492 (2012). 10.1038/nrg 323022641018 · doi ↗ · pubmed ↗
- 2Ong C.T., Corces V.G.: Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 12, 283–293 (2011). 10.1038/nrg 295721358745 PMC 3175006 · doi ↗ · pubmed ↗
- 3Schuff M., Strong A.D., Welborn L.K., Ziermann-Canabarro J.M.: Imprinting as Basis for Complex Evolutionary Novelties in Eutherians. Biology (Basel). 13 (2024). 10.3390/biology 13090682 PMC 1142881339336109 · doi ↗ · pubmed ↗
- 4Gifford C.A., : Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell. 153, 1149–1163 (2013). 10.1016/j.cell.2013.04.03723664763 PMC 3709577 · doi ↗ · pubmed ↗
- 5Xie W., : Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 153, 1134–1148 (2013). 10.1016/j.cell.2013.04.02223664764 PMC 3786220 · doi ↗ · pubmed ↗
- 6Okano M., Bell D.W., Haber D.A., Li E.: DNA methyltransferases Dnmt 3a and Dnmt 3b are essential for de novo methylation and mammalian development. Cell. 99, 247–257 (1999). 10.1016/s 0092-8674(00)81656-610555141 · doi ↗ · pubmed ↗
- 7Bick A.G., : Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. 586, 763–768 (2020). 10.1038/s 41586-020-2819-233057201 PMC 7944936 · doi ↗ · pubmed ↗
- 8Jaiswal S., : Age-related clonal hematopoiesis associated with adverse outcomes. N Engl. J. Med. 371, 2488–2498 (2014). 10.1056/NEJ Moa 140861725426837 PMC 4306669 · doi ↗ · pubmed ↗