Challenging the notion of Aedes aegypti as the primary chikungunya virus vector: insights from Kédougou, Southeastern Senegal
Alioune Gaye, Moussa Moïse Diagne, Diawo Diallo, El Hadji Ndiaye, Marie Henriette Dior Ndione, Moussa Gaye, Idrissa Dieng, Madeleine Dieng, Mouhamed Kane, Safietou Sankhe, Babacar Diouf, Faty Amadou Sy, Caroline Weldon, Ibrahima Dia, Scott C. Weaver, Mawlouth Diallo

TL;DR
This study challenges the belief that Aedes aegypti is the main mosquito spreading chikungunya virus in southeastern Senegal, finding that Ae. furcifer and other species may play a bigger role.
Contribution
The study reveals that Ae. furcifer, not Ae. aegypti, is the most abundant host-seeking species and a major vector of chikungunya virus in the region.
Findings
CHIKV was detected in 31 mosquito pools, with Ae. furcifer being the most infected species.
Aedes aegypti was present but only one pool tested positive for CHIKV.
Anopheles gambiae, the main malaria vector, also tested positive for CHIKV.
Abstract
Chikungunya fever (CHIK) caused by the mosquito-borne chikungunya virus (CHIKV) and transmitted by Aedes mosquitoes, remains a public health burden throughout the tropics. During the CHIK outbreak in the southeastern Senegal in August 2023, an entomologic investigation was conducted to identify the vector(s) and characterize the virus strains. Adult mosquitoes were collected indoors and outdoors from houses of confirmed CHIK cases and their immediate neighborhoods using Prokopack aspirators and double-net traps and all water containers were inspected for aquatic stages. Mosquito pools were tested for CHIKV by RT-qPCR and positive samples were subjected to whole genome sequencing using Illumina iSeq system. Animal watering points; bricks and tree holes were the most common sites for Aedes aegypti larvae and pupae. While immature Ae. Aegypti were found in all affected villages, with…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
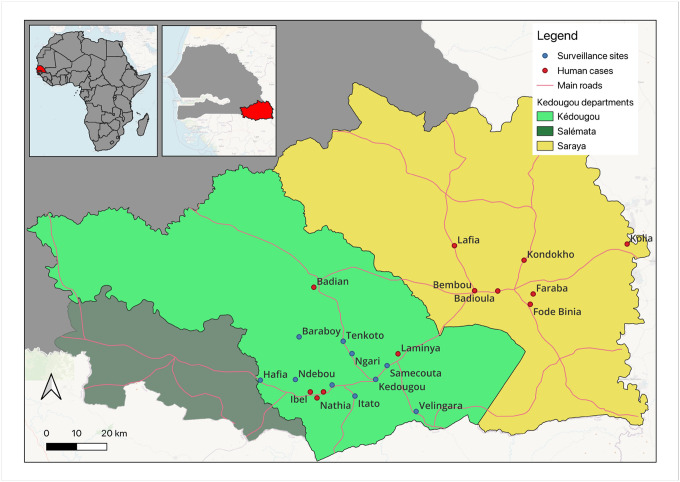 Figure 1
Figure 1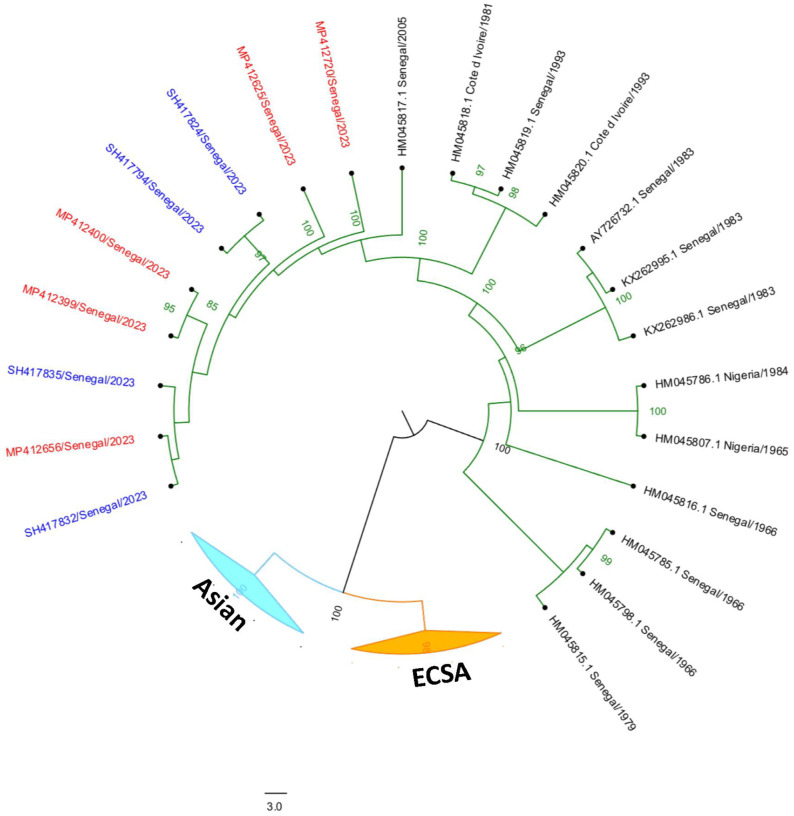 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMosquito-borne diseases and control · Dengue and Mosquito Control Research · Malaria Research and Control
Background
Chikungunya virus (CHIKV) is a mosquito-borne alphavirus of the family Togaviridae, that causes a febrile illness, often accompanied by severe joint pain as well as other common signs and symptoms including muscle pain, headache, nausea, fatigue and rash [1]. The joint pain associated with chikungunya can be debilitating and may persist for weeks or months [2]. Known to be endemic for at least decades in Africa and Asia [3], CHIKV has spread in recent years to other parts of the world, including Europe [4] and the Americas [5]. CHIKV is transmitted to humans by mosquitoes, with Aedes aegypti recognized as the principal epidemic vector in most locations worldwide, and Ae. albopictus as a secondary vector. In Africa sylvatic Aedes mosquitoes are considered to play an important role only in CHIKV maintenance in the enzootic cycle involving non-human primates living in the forest canopies [6, 7].
In West Africa, sylvatic CHIK amplification tends to occur cyclically, approximately every 4 years [8]. As reported in previous publications, an outbreak was detected in 2006 in southeastern Senegal with only 6 confirmed cases [9], coinciding with an outbreak in Nigeria and Cameroon [10, 11]. Subsequently in 2009, another outbreak affected Ghana, Ivory Coast and Burkina Faso, as well as southeastern Senegal, where 20 human cases were confirmed [12, 13]. More recently in 2015, another CHIK outbreak occurred in the Kedougou region of Senegal with 40 confirmed cases reported across its three administrative subdivisions: Kedougou, Salemata and Saraya [14].
Various factors are suspected to influence the cyclic emergence of CHIKV including climatic conditions such as temperature, rainfall and humidity, which impact the vectorial capacity of mosquitoes. Additionally, the herd immunity acquired during previous outbreaks may temporarily reduce the transmission intensity. Over time, CHIKV undergoes genetic changes that can affect its ability to replicate, be transmitted, or evade the host immune response. Moreover, human activities and movement can contribute to virus spread and its introduction into new areas with young, susceptible populations.
In August 2023, Senegal experienced a CHIK outbreak, with over 200 confirmed cases reported in the Kedougou region [15]. An entomological investigation was undertaken with the objective to identify the mosquito vectors involved and genetically characterize the virus responsible of the epidemic.
Materials and Methods
Study site
Mosquito collections were carried out during August to September, 2023, across ten geographic sites in southeastern Senegal, encompassing five predominant land cover classes: village, agriculture, barren, savanna, forest as previously described [6]. In addition, 13 villages (Laminya, Nathia, Boundoucondi, Ibel, Badian, Fodé Binia, Lafia, Bembou, Faraba, Badioula, Kolia and Kondokho) where human chikungunya were confirmed, were investigated. Figure 1 shows villages with human cases (in red) and surveillance sites (in blue) where mosquitoes were sampled, Southeastern Senegal, August 2023. Adult mosquitoes were sampled across all biotopes and localities, while immature stages were sampled exclusively in villages where human cases were detected.
Mosquito sampling
Immature stages sampling was performed indoor and outdoor of randomly selected human habitations. All artificial and natural water-holding containers were inspected, using a flashlight if necessary, and considered as positive when harboring at least one larva or pupa of Ae. aegypti. From each positive container, a sample of larvae and/or pupae was collected, reared to adulthood, and identified morphologically.
Resting and host-seeking adult mosquitoes were collected indoors and outdoors in each locality using a Prokopack aspirator [16]. Additionally, host-seeking mosquitoes were collected in the other landcover classes by aspiration in a double net baited by humans.
Sample processing.
Immature mosquitoes sampled from water container were reared to adulthood, while adults collected in nature were processed directly. Morphological identification of mosquitoes was performed on a chill table using appropriate keys [17–21]. Mosquitoes were pooled by species, sex, physiological status (engorged or unengorged), collection method, location, and date. Pools were then tested in the field laboratory for virus detection. Field-based Next Generation sequencing was also used for whole genome sequencing of positive samples.
Molecular detection.
Mosquito pools were homogenized in 1.5-ml Eppendorf tubes containing 500 μl of L-15 medium (Gibco BRL, Grand Island, NY, USA) using sterile pestles in a biosafety level 2 laboratory. The homogenates were centrifuged at 8,000 rpm for 10 min at 4°C. The supernatant was retained for further analyses, while mosquito debris was discarded.
For CHIKV detection, RNA was extracted from 140 μl of supernatant using the QiaAmp Viral RNA Extraction Kit (Qiagen, Heiden, Germany) following the manufacturer’s protocol. RNA was amplified using a one-step real-time RT-qPCR assay with the TIB Molbiol LightMix^®^ (Berlin, Germany). The 20 μL reaction volume consisted of 5 μL of extracted RNA, 2x Master Mix, 10 μM of specific primers (Forward: AAg CTY CgC gTC CTT TAC CAAg, Reverse: CCA AAT TgT CCY ggT CTT CCT) and probe (6FAM/CCA Atg TCY TCM gCC Tgg ACA CCTTT/TMR) targeting CHIKV in singleplex.
Whole Genome Sequencing and phylogenetic analysis.
Confirmed CHIKV-positive samples were processed for field-based Next Generation sequencing using the Twist Biosciences Comprehensive Viral Research Panel (CVRP) to obtain the whole viral genome. Enriched sample libraries were loaded onto an Illumina iSeq 100 sequencing system, and genome assembly was performed using the Chan Zuckerberg ID (CZ-ID; formerly IDSeq) platform [22].
All generated sequences were aligned using MAFFT with a representative CHIKV dataset covering the West African (WA), East-Central-South African (ECSA) and Asian genotypes. Maximum likelihood trees were generated using IQ-TREE [23] with a 1,000 bootstrap iterations, utilizing the best-fit substitution model determined by ModelFinder and 1,000 bootstrap replicates for statistical reliability and visualization was made with FigTree V1.4.4 [24].
Data analysis
Data from larval surveys were used to estimate three key entomological indices: the Breteau Index (BI) defined as the number of containers positive for immature stages of Ae. aegypti per 100 housing units [25], the Container Index (CI) representing the number of containers positive for immature Ae. aegypti per 100 inspected water containers [26] and the Breeding Preference (BP) defined as the ratio of the percentage of positive containers (Y) to the percentage of that type of inspected container (X). The highest Y/X ratio indicated the preferred type of breeding site for mosquitoes. Epidemic thresholds for BI and CI were set at 5% and 3%, respectively, based on WHO standards [27]. The minimum field infection rates for CHIKV were calculated, including 95% confidence intervals (lower and upper limits), using the PooledInfRate software, version 4.0 [28]. Statistical differences were determined with a significance level set at p < 0.05.
Results
Collection of immature stages and adult mosquitoes.
A total of 742 water containers was inspected across the surveyed sites, showing significant variations in Ae. aegypti breeding preferences between administrative departments. In Saraya tree holes emerged as the most preferred sites, followed by bricks. In contrast, in Kédougou department, animal watering troughs and bricks were the sites most commonly inhabited by mosquito immatures (Table 1). At the village level, animal watering troughs were identified as the most commonly occupied containers in 9 out of 12 villages. Other notable immature habitats included tree-holes, bricks, water storage containers and tires. In all the villages investigated, the epidemic risk indices consistently exceeded the thresholds defined by the WHO across all surveyed villages (see additional file).
A total of 6209 adult mosquitoes belonging to 49 species and 8 genera were collected during the period of the CHIK outbreak. Of these, 31 mosquito pools comprising 8 species (7 Aedes and 1 Anopheles species) were found infected by CHIKV (Table 2). Ae. furcifer was the only species found infected across all biotopes. In the forest biotope, Ae. taylori was the most frequently infected species (p = 0.03, statistically significantly higher than Ae. vittatus,) followed by Ae. furcifer. In the savanna, Ae. dalzieli showed the highest infection rate (p = 0.02, statistically significantly higher than Ae. vittatus,) followed by Ae. furcifer. In village settings, An. gambiae was the most frequently infected species followed by Ae. furcifer, while in Barren and Agriculture environments, Ae. furcifer was the only infected species.
Phylogenetic analysis
The phylogenetic analysis revealed that the 2023 CHIKV outbreak strain in Kedougou belongs to the WA genotype, forming a highly supported monophyletic group (bootstrap ≥ 95) with strains from the 2015 and 2005 outbreaks in the same region [29]. These sequences share 98.80–98.96% nucleotide identity with the reference strain HM045817 (2005), underscoring their close genetic relationship. Furthermore, the clustering of mosquito and human sequences confirmed that the same viral strain circulated between these hosts during the outbreak.
Figure 2 provides results of the phylogenetic analysis of CHIKV sequences obtained from mosquitoes (labeled with red identifier numbers) and humans (labeled with blue identifier numbers) during the 2023 outbreak in southeastern Senegal. The sequences are grouped according to their genetic lineage, including those belonging to the West African, East/Central/South African (ECSA), and Asian genotypes.
Discussion
Nearly worldwide, Aedes aegypti along with Ae. albopictus is considered the primary epidemic vector of several common, human-amplified arboviruses such as dengue, yellow fever, Zika and CHIKV [6, 30–33]. In fact, Ae. aegypti has been several times found associated with CHIKV in central [34], southern [35] and eastern parts of Africa [36], and also identified elsewhere in Africa as the main epidemic vector [37, 38]. Furthermore, populations of Ae. aegypti from South Africa [39], Cameroon [40], Senegal and Cape Verde [41, 42] have been shown to be experimentally competent for CHIKV transmission. Moreover, vertical transmission of CHIKV in Ae. aegypti has been demonstrated experimentally in India [37]. All of these results support a major CHIKV vector role for Ae. aegypti.
For many decades, Ae. aegypti formosus, the ancestral, sylvatic form that evolved into the domesticated Ae. aegypti aegypti, was considered as the only subspecies present in southern Senegal [43]. However, recent studies have shown the presence of both subspecies in Kédougou department [44] with larval development sites found in both forest and, more recently, in domestic environments. These changing distribution patterns are likely linked to deforestation driven by agriculture, gold mining and other human activities, leading to the domestication of wild Ae. aegypti populations in response to urbanization. Such adaptation to the domestic environment has also been observed in Nigeria [45] and Gabon [46].
In our study, Ae. aegypti was abundant in immature stages in all potential aquatic sites and the epidemic risk indices were above the thresholds defined by the WHO [25, 26]. In other words, the indices were sufficiently high for this species to ensure CHIK transmission. Also, at the adult stage Ae. aegypti were found during our investigation in all affected localities. However, CHIKV infection in Ae. aegypti was surprisingly low, with only one positive pool, representing just 3.2% of positives. Therefore, the main epidemic vector worldwide apparently did not play a major role in CHIKV transmission to humans during this outbreak. The zoophilic tendency of Ae. aegypti generally observed in West Africa [47] may explain this finding. The Ae. aegypti formosus form is significantly more abundant in the Kedougou area. However, it has been previously shown to be less endophilic and anthropophilic than the Ae. aegyptiaegypti and thus less involved in human-amplified arbovirus transmission [48]. Therefore, Ae. aegypti formosus probably plays no major role in either maintenance of the sylvatic cycle or the spillover of CHIKV to humans in this region [6].
Our findings highlight a significant involvement of wild mosquitoes in CHIKV transmission during this epidemic, suggesting epizootic amplification of the sylvatic cycle. CHIKV was detected in 29 pools of wild Aedes mosquitoes, predominantly Ae. furcifer (22 pools), which accounted for 71% of positives. Historically, Ae. furcifer has been found most frequently infected with CHIKV [49, 50] and is competent to transmit the virus [51, 52].
The same profile of infected mosquitoes was previously observed in 2009–2010, when among the 42 CHIKV-positive pools, most were wild mosquitoes. Only one pool of Ae. aegypti was found positive [6]. The same trend was observed during the 2015 amplification with the sylvatic vectors recording 93.7% of the CHIKV detections and only 6.25% (2/32 pools) for Ae. aegypti [53]. These findings corroborated our results and support the conclusion that Ae. aegypti does not play a significant role in CHIKV outbreaks in the Kedougou region.
The higher number of human cases (over 200 persons) recorded among men and young people over 15 years of age [15] supports the hypothesis that transmission occurred mainly outside of human dwellings. Sustained domestic transmission would be expected to affect age groups, including children under 15 years of age, who generally stay at home and spend more time indoors. Ae. furcifer, known to have great ecological plasticity [6], was frequently found infected and biting humans in villages at high density in Senegal [6, 43, 54] and elsewhere in Africa [55], indicating its major role in CHIKV transmission.
We also found during our investigation one PCR-positive pool of male An. gambiae. Other species within the Anopheles genus such as An. funestus, An. coustani and An. domicola have been found naturally infected with CHIKV in the Kédougou region during a previous study in 2009–2010 [6]. Subsequently in 2015, 3 pools of female An. gambia ewere found positive for CHIKV in the same area during a co-amplification with YFV and ZIKV [53], suggesting sustained CHIKV transmission by An. gambiae. Vector competence studies of An. gambiae showed a CHIKV infection rate of 43% at day 7 post-bloodmeal, although viral titers in the mosquitoe hemocoel were low and CHIKV-positive saliva was not detected [56]. Moreover An. gambiae and An. funestus, both present in the Kédougou region and transmitting malaria parasites, are the only known vectors of o’nyong-nyong virus (ONNV), a close relative of CHIKV [57, 58]. Also, An. stephensi was recently shown to be a competent ONNV vector [59]. All of these findings suggested that malaria vectors should be monitored for their potential role in CHIKV transmission in Senegal and elsewhere in Africa.
Phylogenetic analysis confirmed that the same CHIKV strain circulated between humans and mosquitoes during 2023, harboring shared mutations compared to CHIKV strains that circulated in 2005 and 2015 [15]. These mutations deserve further study as CHIKV mutations can greatly affect vector host range [60].
Conclusions
This study highlights the persistence of an endemic CHIKV strain in a sylvatic transmission cycle primarily, emphasizing the significant role of wild, sylvatic Aedes species in CHIKV amplification and transmission to humans. Our findings underscore the urgent need to assess the insecticide resistance status of these species and integrate them into vector control programs. Further studies are required to better understand the many factors that contribute to the amplifications of arboviruses like CHIKV at the interface of humans and both sylvatic and urban environments, which can facilitate virus re-emergence and transmission.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Pialoux G, Gaüzère B-A, Jauréguiberry S, Strobel M. Chikungunya, an epidemic arbovirosis. Lancet Infect Dis. 2007;7:319–27.17448935 10.1016/S 1473-3099(07)70107-X · doi ↗ · pubmed ↗
- 2Kularatne SAM, Gihan MC, Weerasinghe SC, Gunasena S. Concurrent outbreaks of Chikungunya and Dengue fever in Kandy, Sri Lanka, 2006–07: a comparative analysis of clinical and laboratory features. Postgrad Med J. 2009;85:342–6.19581242 10.1136/pgmj.2007.066746 · doi ↗ · pubmed ↗
- 3Volk SM, Chen R, Tsetsarkin KA, Adams AP, Garcia TI, Sall AA, Genome-Scale Phylogenetic Analyses of Chikungunya Virus Reveal Independent Emergences of Recent Epidemics and Various Evolutionary Rates. J Virol. 2010;84:6497–504.20410280 10.1128/JVI.01603-09PMC 2903258 · doi ↗ · pubmed ↗
- 4Delisle E, Rousseau C, Broche B, Leparc-Goffart I, L’Ambert G, Cochet A, Chikungunya outbreak in Montpellier, France, September to October 2014. Eurosurveillance. 2015;20.10.2807/1560-7917.es 2015.20.17.2110825955774 · doi ↗ · pubmed ↗
- 5Leparc-Goffart I, Nougairede A, Cassadou S, Prat C, De Lamballerie X. Chikungunya in the Americas. The Lancet. 2014;383:514.10.1016/S 0140-6736(14)60185-924506907 · doi ↗ · pubmed ↗
- 6Diallo D, Sall AA, Buenemann M, Chen R, Faye O, Diagne CT, Landscape ecology of sylvatic chikungunya virus and mosquito vectors in southeastern Senegal. P Lo S Negl Trop Dis. 2012;6:e 1649.22720097 10.1371/journal.pntd.0001649 PMC 3373654 · doi ↗ · pubmed ↗
- 7Russo G, Subissi L, Rezza G. Chikungunya fever in Africa: a systematic review. Pathog Glob Health. 2020;114:136–44.32308158 10.1080/20477724.2020.1748965 PMC 7241529 · doi ↗ · pubmed ↗
- 8Althouse BM, Hanley KA, Diallo M, Sall AA, Ba Y, Faye O, Impact of climate and mosquito vector abundance on sylvatic arbovirus circulation dynamics in Senegal. Am J Trop Med Hyg. 2015;92:88.25404071 10.4269/ajtmh.13-0617 PMC 4347398 · doi ↗ · pubmed ↗