The pancreatic β-cell incretin response is modulated by mitochondrial transaminase GPT2
Sabyasachi Sen, Andrea V. Rozo, Matthew W. Haemmerle, Jeffrey Roman, Andrea V. Scota, Xiaodun Yang, Christine A. Juliana, Sarah A. Tersey, Eric M. Morrow, Nicolai M. Doliba, Doris A. Stoffers

TL;DR
This study shows that reducing GPT2 improves β-cell function and incretin response in type 2 diabetes, suggesting it as a potential therapeutic target.
Contribution
The study identifies GPT2 as a novel modulator of β-cell incretin sensitivity and function in type 2 diabetes.
Findings
Gpt2βKO mice showed improved oral glucose tolerance and insulin secretion.
GPT2 was increased in human islets from T2D donors and under glucolipotoxic conditions.
Silencing GPT2 reversed incretin unresponsiveness in T2D islets and improved β-cell survival.
Abstract
The effect of the incretin hormones GLP-1 and GIP to promote pancreatic β-cell function is exploited by an expansive menu of incretin mimetics for the treatment of type 2 diabetes (T2D); however, the incretin effect is well known to diminish as T2D progresses. Here, we show that silencing of stress-inducible mitochondrial protein glutamic pyruvate transaminase 2 (GPT2) enhances the β-cell incretin response. Mice with β-cell specific Gpt2 deficiency (Gpt2βKO) have improved oral glucose tolerance and insulin secretion due to enhanced β-cell incretin sensitivity. In the diet induced obesity (DIO) model of T2D, Gpt2βKO mice maintained lower non-fasting glucose and improved oral glucose tolerance and insulin secretion. The effect of GLP-1 receptor (GLP-1R) agonism on β-cell survival was also enhanced in Gpt2βKO islets. GPT2 was markedly induced in human islets from donors with type 2…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
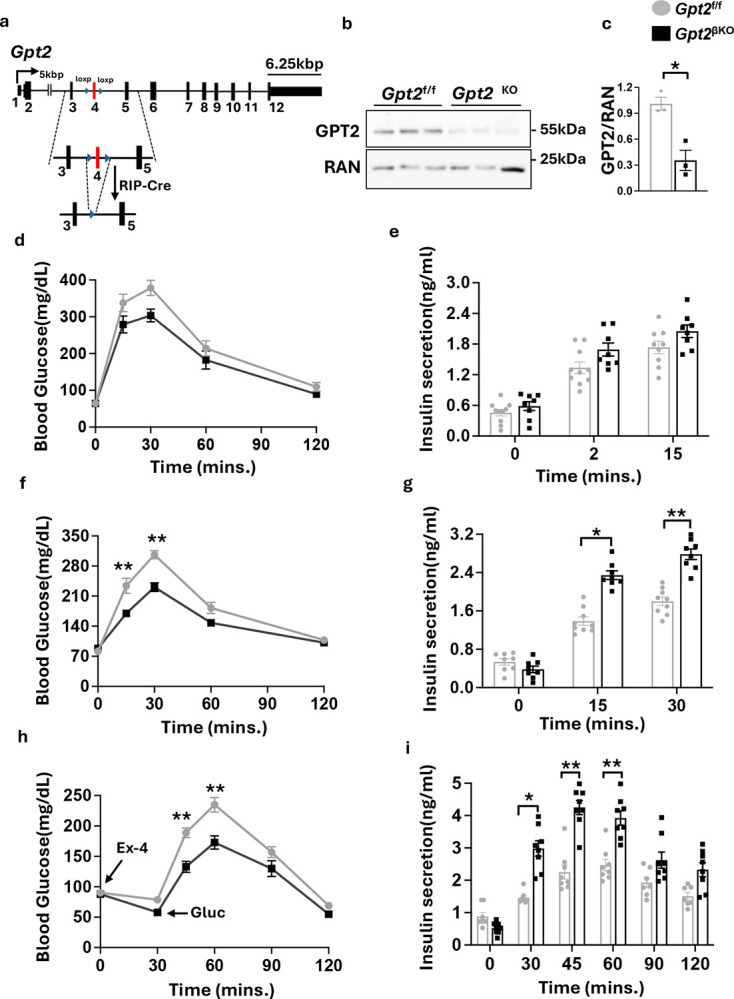 Figure 1
Figure 1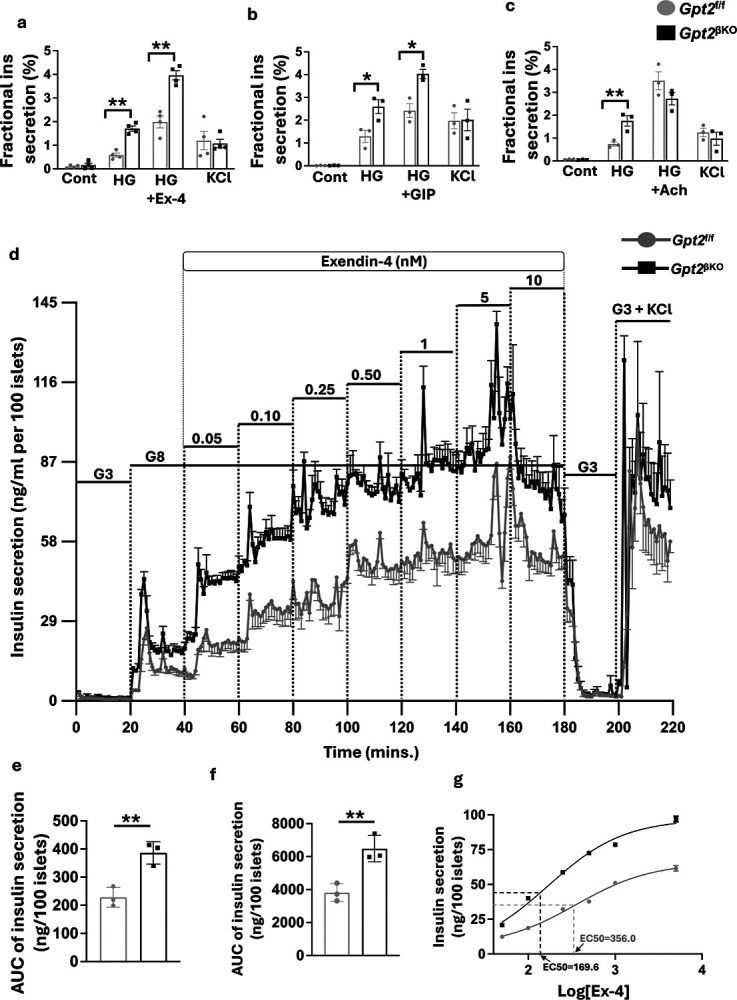 Figure 2
Figure 2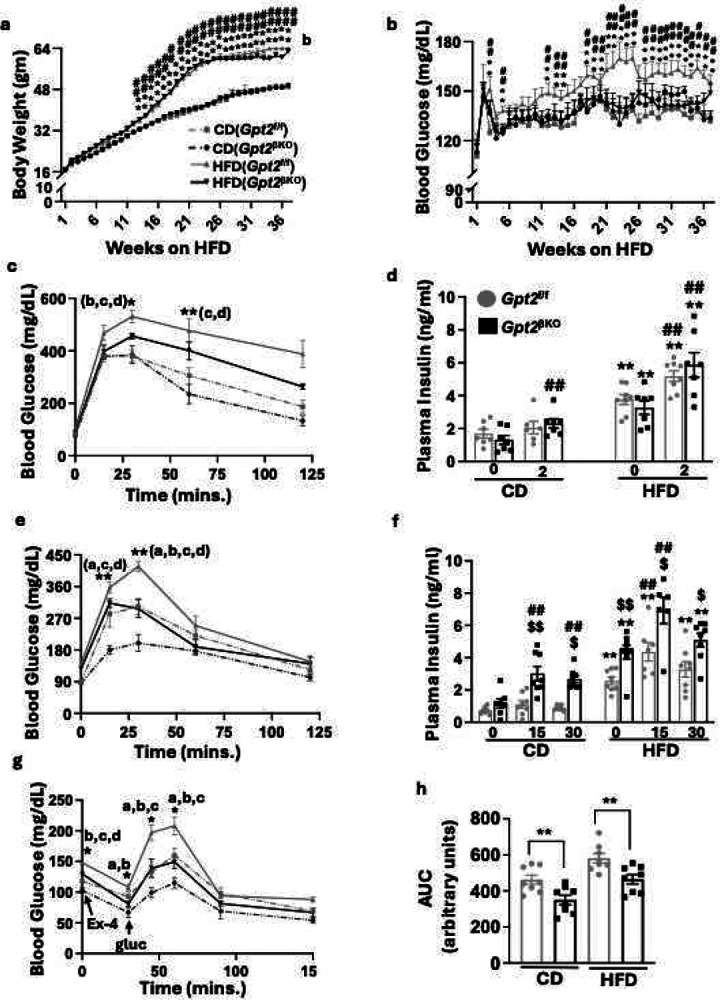 Figure 3
Figure 3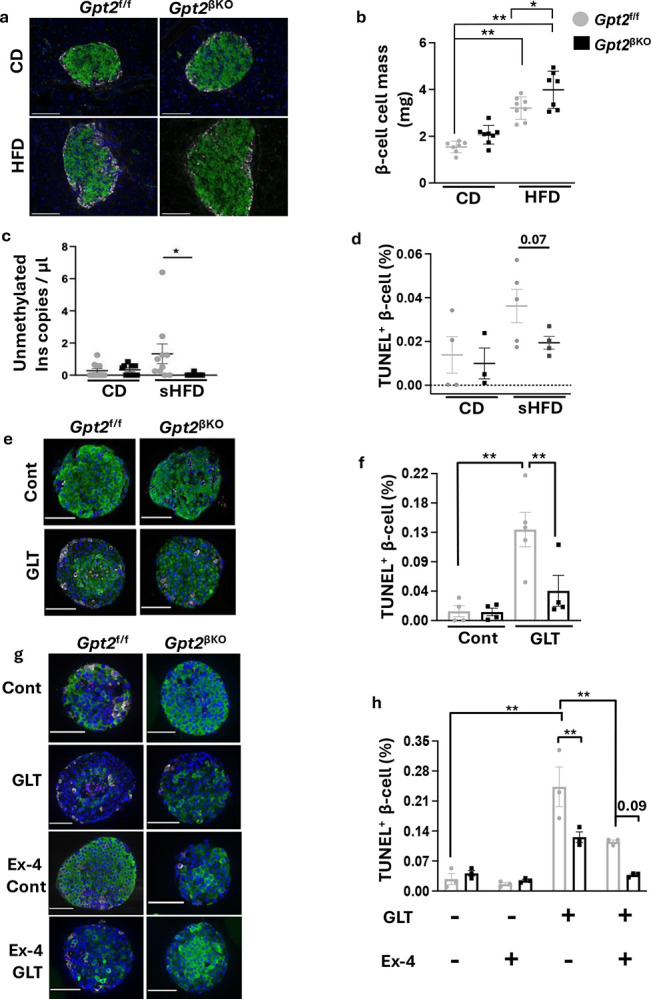 Figure 4
Figure 4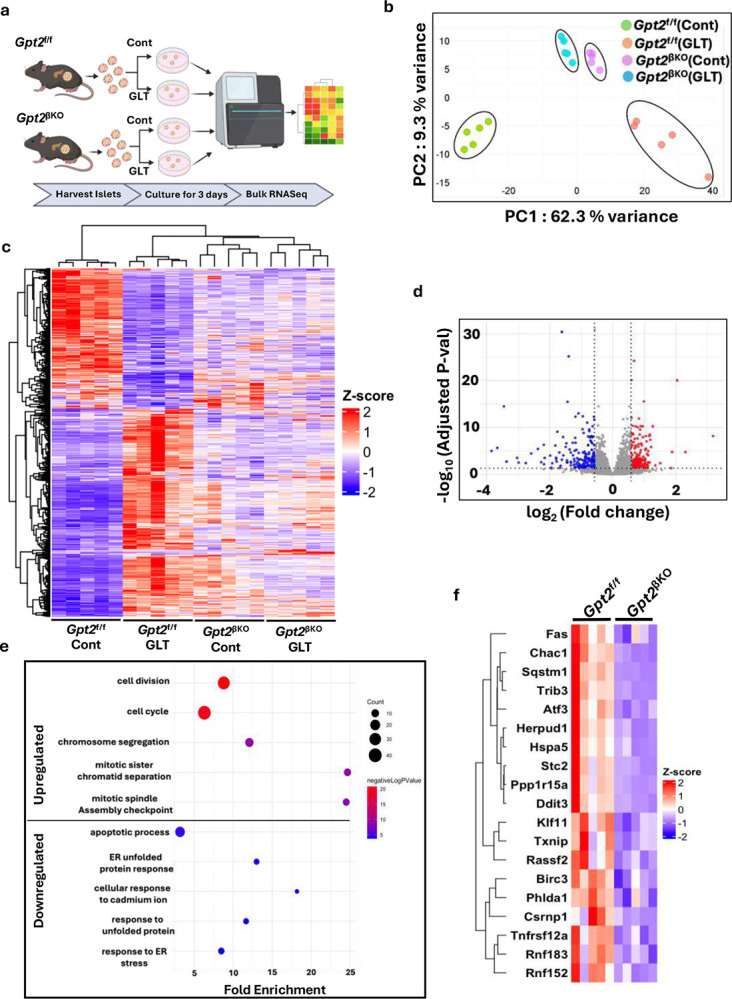 Figure 5
Figure 5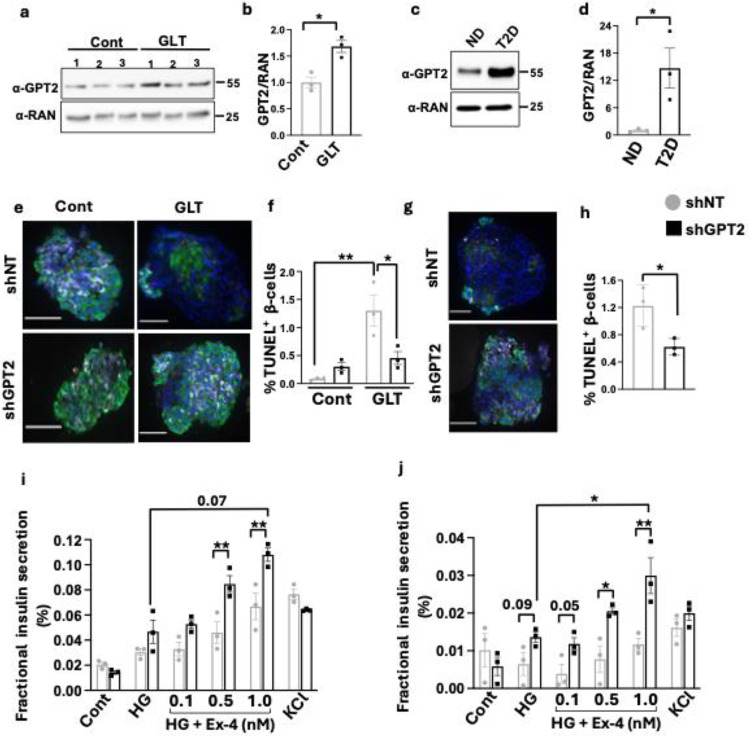 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPancreatic function and diabetes · Diabetes Management and Research · Diabetes and associated disorders
Introduction
The actions of two gut derived polypeptides: GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1), commonly referred to as ‘incretin’ hormones^1^, are responsible for more than 50% of postprandial insulin output from pancreatic β-cells^2–5^. These peptides act on β-cells through their cognate G protein–coupled receptor GPCRs to potentiate glucose-stimulated insulin secretion while simultaneously promoting β-cell survival and replication^6–8^. Progressive loss of functional β-cell mass^9–11^ and decline in incretin sensitivity^12–14^ are central to pancreatic β-cell failure in type 2 diabetes (T2D). Hyperglycemia and high levels of circulating free fatty-acid have been shown to promote β-cell apoptosis and contribute to loss of incretin sensitivity^15–17^. GLP-1R agonists exploit this physiology to counter the effects of glucolipotoxicity (GLT) and improve beta cell function and survival^18–20^. However, the molecular mechanisms underlying these effects remains poorly understood^21^.
We previously identified GPT2 as a stress-induced gene in both human and mouse islets whose silencing in mouse β-cells ex vivo protects from ER stress induced apoptosis^22^. GPT2 encodes a mitochondrial aminotransferase that catalyzes the bidirectional conversion of pyruvate and glutamate to alanine and a-ketoglutarate. It has been implicated in glutamine mediated TCA-cycle anaplerosis in several cancers^23–25^ and primary neurons^26–28^.
Here, we find that GPT2 modulates the sensitivity of both mouse and human β cells to incretin hormones. Mice with β-cell-specific Gpt2 deficiency exhibit enhanced incretin stimulated insulin secretion and cell survival when maintained on regular chow and during high fat diet feeding. GPT2 expression is markedly upregulated in islets from donors with T2D and induced in non-diabetic human islets exposed to GLT. shRNA mediated silencing of GPT2 in the β-cells of human islets from non-diabetic donors enhances sensitivity to the GLP-1R agonist Exendin-4 (Ex4) and alleviates GLT induced apoptosis. Of high translational relevance, silencing of GPT2 in the β-cells of human T2D islets restored glucose and Ex4 responsiveness, raising GPT2 as a therapeutic target to mitigate β-cell dysfunction in diabetes.
Results
Heightened response of Gpt2βKO mice to the GLP-1 receptor agonist Exendin-4
We previously identified mitochondrial transaminase Gpt2 as a transcriptional target of a stress-inducible PDX1-ATF transcriptional complex. Stress upregulation and the presence of a conserved PDX-1 binding CARE motif near the human GPT2 locus suggested conservation of this regulatory network across species. Moreover, silencing Gpt2 in primary mouse islets exposed to thapsigargin or glucolipotoxic stress markedly reduced β-cell apoptosis^22,29^. Here, we set out to investigate the physiological repercussions of Gpt2 depletion in pancreatic β-cells in vivo. We generated a Gpt2 deficiency mouse model (Gpt2^βKO^), with Gpt2^f/f^ mice as controls (Fig.1a). Efficient deletion was verified by western blot analysis (Fig.1b, c). A mild improvement in glucose tolerance and insulin secretion in response to intraperitoneal glucose challenge was observed in Gpt2^βKO^ mice compared to Gpt2^f/f^ littermates (Fig1d and 1e, respectively). In contrast, when challenged with oral glucose, Gpt2^βKO^ mice had significantly improved glucose tolerance and higher insulin secretion than control littermates (Fig.1f, g). Minimal phenotypes were observed in female Gpt2^βKO^ mice (Supplementary Fig.1).
The preferentially higher response of Gpt2^βKO^ mice to oral glucose administration suggested an enhanced incretin effect. GLP1 and GIP, the two major incretin hormones, account for 50–70% of insulin secreted in healthy individuals after meal intake. GLP-1 and GIP have also been implicated in the suppression of β-cell apoptosis, increased β-cell mass and the amelioration of glucotoxic effects during diabetes^30–32^, and loss of incretin sensitivity, a hallmark of type-2 diabetes^12^. To assess beta cell sensitivity to incretin hormones, we injected a low dose of the dipeptidyl peptidase 4 (DPP4) resistant GLP1R agonist, Exendin-4 (Ex4) (0.008ng/kg body weight)^33,34^ to male Gpt2^f/f^ and Gpt2^βKO^ mice and performed an intraperitoneal glucose tolerance test (IPGTT) 30 mins later. Following Ex4, Gpt2^βKO^ mice showed greater initial reduction in blood glucose as well as lower blood glucose excursion compared to Gpt2^f/f^ littermates (Fig.1h). Concomitantly, Gpt2^βKO^ mice also secreted significantly more insulin at 30-, 45-, and 60-minutes after Ex4, suggesting a heightened β-cell response to the GLP-1R agonist (Fig.1i).
Improved incretin sensitivity of Gpt2βKO mouse islets
In addition to the well-established direct effects on pancreatic islet β-cells, GLP1R agonists exert pleiotropic effects on peripheral tissues, including liver, brain and gut, which can in turn influence β-cell function. Therefore, we investigated if our in vivo observations could be recapitulated in isolated islets under ex vivo conditions. Isolated Gpt2^f/f^ control and Gpt2^βKO^ islets were incubated with basal glucose (2.8mM), followed by high glucose (30mM), and then high glucose with one of 3 different insulin secretogogues: Ex4 (1nM), GIP (1nM), or Acetylcholine (Ach; 0.5uM), and finally with KCl (30mM KCl in 2.8mM glucose) (Fig.2a,b, and c respectively). Isolated islets from Gpt2^βKO^ mice had higher insulin secretion in response to high glucose in all three cases, indicating that β-cell depletion of Gpt2 enhances glucose responsiveness. Interestingly, Gpt2^βKO^ islets had markedly higher insulin secretion in response to Ex4 and GIP, but not acetylcholine. To ascertain if Gpt2 depletion enhances islet sensitivity to incretin hormones, we conducted a dynamic perifusion assay wherein Gpt2^f/f^ and Gpt2^βKO^ islets were exposed to low glucose (3mM), high glucose (8mM), and stepwise increments of Ex4 (50pM −10nM) in the presence of 8mM glucose (Fig.2d). Gpt2^βKO^ islets had a higher response to 8mM glucose and markedly enhanced insulin secretion during stepwise addition of Ex4 and a lower EC50 (169.6 nM for Gpt2^βKO^ islets vs 356 nM for Gpt2^f/f^ islets), indicating greater sensitivity to Ex4 (Fig. 2e–g). Taken together, these results indicate that the augmented incretin responsiveness observed in Gpt2^βKO^ mice represents an islet-autonomous phenomenon in which Gpt2 depleted β-cells are more sensitive to the GLP-1R agonist Ex4.
Enhanced incretin effect in Gpt2βKO mice during HFD-feeding
Loss of β-cell incretin sensitivity is a hallmark of T2D, contributing to impaired glucose homeostasis^4,35^. In mice, metabolic stress induced by high calorie diet has been reported to mimic the diabetic milieu and negatively impact the incretin response by lowering β-cell sensitivity to incretin hormones^36^. We, therefore assessed the impact of prolonged high fat (HFD, 45 kcal%) or matched control diet (CD) feeding on Gpt2^βKO^ and Gpt2^f/f^ mice. During the 37 weeks of diet intervention, the Gpt2^βKO^-HFD group showed markedly lower non-fasting, weekly blood glucose levels than the Gpt2^f/f^-HFD group (Fig.3b), despite similar weight gain (Fig.3a). Insulin tolerance was unchanged between the genotypes on either CD or HFD, indicating that changes in glucose tolerance are likely due to improvement in β-cell function (Supplementary Fig.2c). Surprisingly, assessment of the glycemic profile following intraperitoneal injection of glucose showed only mild improvement in glucose tolerance (Fig.3c) and no improvement in insulin secretion (Fig.3d). In striking contrast, following oral glucose injection, the Gpt2^βKO^ mice showed marked improvements in both glucose tolerance (Fig.3e) and insulin secretion (Fig.3f). Circulating levels of the two major incretin hormones, GLP-1 and GIP, were unaltered (Supplementary Fig. 2a and b, respectively), suggesting that enhanced incretin sensitivity of Gpt2 depleted β-cells is responsible for the improved β-cell function. To specifically examine the effect of Gpt2 in modulating β-cell incretin sensitivity, we injected Gpt2^βKO^ and Gpt2^f/f^ littermates with Ex4 and performed an IPGTT 30 minutes later. Gpt2^βKO^ mice showed an improved glycemic profile and markedly higher insulin secretion after Ex4, consistent with our previous observations in chow fed mice (Fig. 3g and 3h). Thus, depletion of Gpt2 in β-cells enhances incretin sensitivity of HFD fed mice, leading to improved glucose homeostasis.
β-cell Gpt2 deficiency enhances the pro-survival effect of Ex4
β-cell mass was increased in Gpt2^βKO^ mice compared to littermate control mice after long term HFD feeding (Fig. 4a, b). The number of TUNEL^+^ β-cells at end of the study was not elevated; however, HFD-induced β-cell death is episodic, and apoptotic cells are rapidly cleared, thus limiting detection^37–39^. Postnatal β-cell mass expansion in rodents occurs by replication of pre-existing β-cells^40,41^. Hence, to determine whether enhanced survival of β-cells early during HFD feeding preserves a pool of functional β-cells allowing for later compensatory expansion of β-cell mass, we assessed β-cell death in vivo in 5-week-old mice after 7 days of HFD-feeding by digital droplet PCR of circulating unmethylated copies of insulin DNA and by TUNEL staining of pancreatic sections at the end of the diet intervention. After 1-week HFD feeding, elevated circulating unmethylated copies of Ins DNA (Fig. 4c) and TUNEL^+^ β-cells (Fig. 4d) were observed in Gpt2^f/f^ mice, and these effects were mitigated in Gpt2^βKO^ mice, indicating a protection from metabolic stress induced β cell death conferred by β cell Gpt2 deficiency.
The protection of β-cell survival by Gpt2 deficiency was recapitulated ex vivo. Isolated Gpt2^f/f^ or Gpt2^βKO^ islets were maintained under control or GLT conditions for 3 days. Gpt2 deletion attenuated beta cell death during GLT, evidenced by a more than three-fold reduction in the percentage of TUNEL^+^ β-cells (Fig. 4e, f). Finally, we assessed the impact of Ex4 on GLT stress-induced apoptosis of Gpt2^f/f^ and Gpt2^βKO^ β-cells. Ex4 lowered TUNEL^+^ β-cells in Gpt2^f/f^ islets, pointing to a protective effect rendered by incretin activation during GLT stress. Remarkably, the combination of Gpt2 deficiency and Ex4 lowered death to control levels, strongly suggesting that Gpt2 silencing augments the pro-survival action of Ex4 during GLT stress (Fig. 4g and 4h).
β-cell Gpt2 silencing counters the transcriptional effects of glucolipotoxicity
To understand the global impact of Gpt2 depletion on biological processes and pathways, we performed bulk RNAseq analysis of Gpt2^f/f^ and Gpt2^βKO^ islets cultured under control or GLT conditions for 3 days (Fig.5a). We observed a significant stratification by genotype and culture condition, indicating substantial differences in gene expression profiles and underlying biological pathways among these conditions (Fig. 5b). Expression levels of significantly altered genes are depicted as hierarchical clustering in a heatmap across sample groups (Fig.5c).
β-cell damage in patients with T2D is often attributed to elevated levels of circulating glucose and lipid^42,43^; hence, multiple studies have used the ex vivo GLT model in islet cultures to replicate the metabolic milieu of T2D^16,44^. To understand how closely ex vivo GLT stress recapitulates T2D, we compared the impact of GLT on gene expression of control (Gpt2^f/f^) islets to two publicly available bulk RNAseq datasets of human islets from non-diabetic donors or donors with T2D: (T2D_1^45^ and T2D_2^46^) We also compared these data with publicly available bulk RNAseq data of db/db mouse islets^47^ which phenocopy the β-cell dysfunction prevalent in T2D islets^48^. We observed significant overlap with gene expression of T2D and db/db islets, indicating that GLT exposed Gpt2^f/f^ islets show congruency in the transcriptional landscape and thus similarity in underlying molecular mechanisms with pathophysiologically relevant β-cell models (Supplementary Fig. 3a).
We next focused on differential gene expression between GLT-treated Gpt2^βKO^ and Gpt2^f/f^ islets to assess how Gpt2 silencing influences the islet response to GLT stress. We found 352 upregulated and 442 downregulated genes (Fig. 5d). The five most significant biological processes enriched in the downregulated gene group were related to ER-stress and apoptosis, whereas the up-regulated gene group highlighted pro-survival programs, such as cell-division and cell-cycle regulation (Fig. 5e). Heatmaps of a key set of genes from ER-stress and apoptosis (Fig. 5f) and from cell-cycle and cell-division pathways (Supplementary Fig. 3b) are shown. We also observed upregulation of key cAMP pathway factors (Crem, Irs2) along with cell-cycle and IGF related pathways in Gpt2^βKO^ versus Gpt2^f/f^ islets under control conditions (Supplementary Fig.3c and 3d). Gpt2 silencing under GLT conditions mirrored previously reported transcriptional effects of Ex4, including lower expression of the ER-stress and pro-apoptotic genes Fas^49^ and Ddit3^50^ and upregulation of the proliferative gene Ccna2^51^. Together, these transcriptional changes support our physiological observations that Gpt2 depletion primes β-cells for enhanced incretin signaling while simultaneously activating intrinsic survival pathways.
GPT2 silencing lowers apoptosis and restores incretin responsiveness of human T2D β-cells
To establish the potential translational relevance of these findings to human disease, we analyzed human islets from non-diabetic donors and from donors with T2D. Similar to mouse islets, GPT2 was induced by GLT in human islets (Fig.6a, b). GPT2 was also markedly increased in islets from T2D donors (Fig.6c, d, Supplementary Fig. 4a). To determine whether GPT2 deletion protects human β-cells from GLT stress-induced apoptosis, we transduced islets from non-diabetic donors with lentivirus directing human β-cell expression of shNT or shGPT2 and exposed them to control or GLT stress conditions. In the transduced islets, 66–88 % insulin-positive β-cells were EGFP positive, confirming high β-cell-specificity for both shRNA constructs (Supplementary Fig. 5c). We observed a significant increase in apoptotic β-cells under GLT stress in these islets, and this was remarkably counteracted by depletion of GPT2 (Fig.6e–f). Importantly, GPT2 silencing in the β-cells of islets from T2D donors significantly reduced the number of apoptotic β-cells (Fig.6g–h).
Incretin responsiveness is impaired in islets from patients with T2D^5,52,53^. To determine the impact of GPT2 silencing on incretin sensitivity of human β-cells, islets from a single, non-diabetic donor were again transduced with shNT or shGPT2 lentivirus, and insulin secretion was assessed after static incubation with basal glucose (2.8mM), high glucose (8mM), and increasing doses of Ex4 in the setting of high glucose (Fig. 6i). To ensure that transduction itself did not alter glucose and Ex4 responsiveness of donor islets, we performed static incubation with un-transduced non-diabetic islets under the same conditions and observed no difference with shNT-transduced islets (Supplementary Fig.4e). GPT2 silencing enhanced Ex4 stimulated insulin secretion, suggesting increased GLP1 sensitivity on reduction of GPT2. We further tested the incretin responsiveness of type 2 diabetic islets after GPT2 silencing and observed improved secretion in response to high glucose and enhancement of Ex4 stimulated insulin secretion, suggesting that GPT2 deletion can restore incretin sensitivity and beta cell function in islets from T2D donors (Fig. 6j).
Discussion
Pancreatic β-cells integrate metabolic and hormonal signals to regulate insulin output^54–56^. Incretin hormones are critical not just for regulating insulin secretion, but also for the survival and replication of β-cells^2,3,57,58^. Despite inherent differences in modes of β-cell incretin activity between murine and human models^59,60^, we present GPT2 as a key factor regulating β-cell incretin activity and survival across species. Gpt2^βKO^ mice exhibited improved incretin sensitivity and enhanced β-cell mass expansion under HFD due to improved β-cell survival. These findings were corroborated in human islets as well, where GPT2 was induced in the context of T2D and in non-diabetic islets exposed to GLT conditions
This work thus establishes a link between glutamine metabolism and incretin stimulated insulin secretion in the beta cell. Glutamine metabolism plays an important role in insulin secretion^61,62^, and it has been suggested, based on experiments in β-cell lines, that glutamate transport into secretory vesicles promotes insulin granule recruitment to the plasma membrane and is thus linked to incretin-stimulated insulin secretion^63^; however, based on the pattern of KCl-stimulated insulin secretion during our GSIS assays, Gpt2 appears to affect mechanisms further upstream. This is additionally supported by the lack of impact of β-cell Gpt2 deficiency on Ach-stimulated secretion. Further investigation of the intersection of glutamine metabolism with incretin hormone signaling in the β cell and the relationship of these pathways to insulin secretion and cell survival is warranted.
Gpt2 silencing in β-cells also amplifies the pro-survival effect of the GLP1R agonist Ex4. Previous studies point towards pro-proliferative and anti-apoptotic roles of incretin signaling in β-cells, linked to upregulation of genes like Ccna2^51^ and Irs2^64,65^ and to downregulation of Fas and Ddit3. Our transcriptomic analysis revealed upregulation of Ccna2 and downregulation of Fas and Ddit3 by Gpt2 silencing during GLT stress whereas key effectors of GLP1R signaling in β-cells like Crem^66,67^ and Irs2 were upregulated by Gpt2 silencing under control conditions, altogether demonstrating that Gpt2 silencing counters the effects of glucolipotoxicity and providing pathways through which Gpt2 silencing promotes Ex4 effects on β cell function and survival.
A growing body of studies on incretins highlights their dual benefit to enhance insulin secretion and preserve β-cell mass. Our study suggests that selective targeting of GPT2 could amplify the therapeutic benefit of GLP-1R agonists to optimize glycemic control. Thus, our findings position GPT2 as a promising therapeutic target for enhancing β-cell lifespan and function during diabetes.
Methods
Generation of conditional Gpt2 deficiency in mice
To generate β-cell specific deletion of Gpt2 in mice (Gpt2^βKO^), we intercrossed C57BL/6 mice with loxP sites flanking exon 4 of Gpt2 (Gpt2^f/f^)^26^ with a β cell specific Cre recombinase deleter strain (Tg(Ins2-Cre)^Herr^)^68^. This deleter strain is characterized by high efficiency targeting of β cells, absence of a GH expressing mini-cassette, minimal brain leakinesss and long history of effective targeting without silencing^69^. We previously documented normal glucose homeostasis in male and female Tg(Ins2-Cre)^Herr^ mice^70,71^. Gpt2^f/f^ littermates served as controls. All animals were housed in the animal care facility of the University of Pennsylvania and maintained on a 12-hour light/dark cycle, with ad libitum access to food and water. Unless otherwise indicated, experiments were performed on male mice.
In vivo metabolic phenotyping
Mice were fasted overnight before administration of 2g/kg BW glucose either by intraperitoneal injection or by oral gavage for glucose tolerance testing (GTT) as previously described^70–72^. Exendin-4 (Ex4; Millipore Sigma, E7144) was reconstituted in sterile 0.9% NaCl, 1% BSA and injected intraperitoneally. Intraperitoneal GTT or glucose stimulated insulin secretion (GSIS) was assessed 30 minutes later. Insulin tolerance testing (ITT) was performed by injecting 0.75U/kg BW insulin (Novolin, 0169–18341) after a 6 hour fast. For both IP- and OGSIS, 3g/kg BW glucose was injected either intraperitoneally or administered by oral gavage. For glucose and insulin measurements, tail vein blood was collected into EDTA coated collection vials (Sarstedt, 16.444.100) followed by plasma separation by centrifugation and storage at −80°C. Insulin was assayed by insulin ELISA (Crystal Chem, 90082) following manufacturers protocol. For measurement of GLP-1 and GIP, Gpt2^f/f^ and Gpt2^βKO^ mice were administered 3g/kg BW glucose by oral gavage and plasma collected from the tail vein was separated as described above. GLP-1and GIP were assessed by ELISA (Meso Scale diagnostics, K1503PD-1 and Crystal Chem, 81527).
For high fat diet (HFD) feeding, 4-week-old male Gpt2^f/f^ and Gpt2^βKO^ mice were weaned onto either a HFD (45% Kcal fat, Research Diets: D12451) or matched control diet (10% Kcal fat, Research Diets: D12450H) for 1 or 37 weeks. Weekly, non-fasting body weight and blood glucose were measured. Blood glucose was measured from a nick in the tail vein by handheld glucometer.
Isolation and culture of human and mouse pancreatic islets.
Primary mouse islets were isolated as previously described^70^ and recovered for 3 days in RPMI 1640 medium supplemented with 11 mM glucose and 10% FBS. De-identified human islets from non-diabetic donors or donors with T2D were obtained through the Integrated Islet Distribution Program or the Human Pancreas Analysis Program, both NIH-approved centers with informed consent and IRB approval. Donor islet characteristics are provided in Supplementary Table 4. For glucolipotoxicity experiments, mouse and non-diabetic human islets were incubated with either control (RPMI,1% BSA) or GLT media containing RPMI supplemented to 25mM glucose and with 0.5 mM FFA (2:1 mix of palmitate and oleate) and 1% BSA for 3 days after recovery^73^.
Cell culture and shRNA preparation
HEK293T cells were cultured in DMEM (Gibco, 11965084) with 10% FBS (GeminiBio, S11150) and 1% penicillin-streptomycin (Thermo Fischer, 15140122). For lipotoxic stress treatment, cells were incubated in the indicated concentrations of FFA with 1% BSA in culture media, with 1% BSA serving as control.
For shRNA mediated silencing in human islets, shRNA sequences targeting the human GPT2 transcript (ENST00000340124.9) were designed using the Sherwood shRNA design algorithm^74^ and 3 shRNAs targeting different regions of the GPT2 transcript were individually cloned into a pLenti plasmid vector that contains CMV promoter driving expression of EGFP with an shRNA acceptor sequence in its 3’UTR^70^. Plasmids were transfected into HEK293T cells using lipofectamine 2000 to assess silencing efficiency.
To create a human β-cell specific shRNA expression system, we first used Gibson-assembly to introduce a RIP1-miniCMV enhancer-promoter that confers >90% high human β cell specificity^75^ into our pLenti EGFP construct. The GPT2 shRNA construct showing highest silencing efficiency in HEK293T cells was cloned into the 3’UTR shRNA acceptor cassette downstream of the EGFP coding sequence and packaged into lentiviral particles. Lentiviral packaging vectors (psPax2 and PMD2G) were transfected to HEK293 cells together with our pLenti-shRNA constructs. Lentiviral particles in the culture media were collected at 24-, 48-, and 72-hours using Lenti-X Concentrator (631232, Takara Biosciences) following manufacturer protocol and resuspended in culture medium before being aliquoted and stored in −80°C. Human islets were transduced with lentivirus media overnight in serum-free media as described previously. To assess β-cell-specificity, paraffin sections of donor islets from non-diabetic individuals transduced with non-targeting (shNT) or GPT2 (shGPT2) shRNAs were stained with anti-insulin, -glucagon or -EGFP antisera. Sequence of the shRNAs tested will be made available upon request.
Immunohistochemistry and Immunofluorescence
Harvested pancreata were weighed before overnight fixation in 4% paraformaldehyde at 4°C followed by paraffin embedding and sectioning. For β-cell mass measurement, 3 sections per animal taken at ~ 200μm intervals in the region of maximal footprint were stained with guinea pig anti-insulin antiserum (Dako), mouse anti-glucagon antiserum (Millipore) and counterstained with Hoechst (Sigma). Antigen retrieval was performed using 20ug/ml Proteinase K in 10mM Tris-HCl, pH-8.
Mouse and human islets were fixed in 4% paraformaldehyde for 30 minutes at room temperature, followed by resuspension in warm histogel and shaping into small discs. Histogel discs were paraffin embedded and sectioned as described previously^76^. Paraffin slides were treated with 10mM sodium citric acid buffer pH 6 with 0.2% Tween20 in a pressure cooker for 2 hours and stained with primary antisera to goat anti-GFP, insulin and glucagon. For β-cell specific shRNA expression in human islets, EGFP expression was used as marker for transduction efficiency and specificity. To assess β-cell death, the In-situ Cell Death Detection kit (Fluorescein; Millipore Sigma, 11684795910) was used following manufacturer’s instructions along with fluorescent conjugated dUTP (Invitrogen A32762). TUNEL+ β-cell number was normalized to total β-cell number.
Images were captured on a QImaging Q/Click digital camera equipped Nikon Eclipse E600 microscope using MetaMorph software. For whole slide scanning at 20X magnification, we used an Aperio Versa 200 slide scanner and images were analyzed using Aperio Imagescope software (Leica biosystems). For each animal, the fraction of total insulin-positive area/total footprint area was multiplied by the respective pancreas wet weight to determine beta cell mass.
For isolated islet sections, scanning was performed with an Agilent Biotek Cytation5 Imaging reader using a 20X/0.45NA Plan-Fluorite phase objective. Captured images were analyzed using Qupath image analysis software (version 0.5.1)^77,78^. Details of primary and secondary antisera used for immunofluorescence are provided in Supplementary Table 3.
Static and dynamic insulin secretion assays
For static incubation studies using mouse or human islets, 25–30 islets were washed and incubated in Krebs-Ringer bicarbonate buffer (KRBB), supplemented with the indicated concentrations of glucose, Ex4, GIP (Fisher Scientific, NC9796977), Acetylcholine (Millipore Sigma, A2661), and potassium chloride (KCl) for 45 minutes each as previously described^70,71^. At the end of the experiment, the islets were extracted for later measurement of insulin content. Insulin content and insulin secreted into the medium for each condition was measured by insulin ELISA (Crystal Chem, for mouse (90082) and human (90115) insulin). Secreted insulin was normalized by insulin content to calculate fractional insulin secretion.
Dynamic perifusion assays were carried out with a Biorep Perifusion PER-04 device. One hundred islets from Gpt2^f/f^ or Gpt2^βKO^ were perifused at 100μl/min with low glucose (G3, 3mM), high glucose (G8, 8mM), followed by stepwise increments of Ex4 (50pM-10nM) in G8 and followed by washout in low glucose and 30mM KCl. Perifusates were collected every minute and assayed in triplicate for insulin by the Radioimmunoassay and Biomarker Core of the Penn Diabetes Research Center.
Digital droplet PCR (ddPCR) for unmethylated Ins DNA
At the end of the 1-week HFD study, blood was collected by cardiac puncture from mice under isoflurane inhalation anesthesia (1.5% v/v O_2_), and serum obtained after centrifugation was stored at −80°C. ddPCR for quantification of differentially methylated Ins gene promoter DNA was carried out as described^79^.
RNA sequencing and data analysis
At least 150 islets per genotype and condition were resuspended in TRIzol, RNA was isolated, and sequencing libraries were prepared using the Illumina Ultra Low Input RNA kit. Libraries were then sequenced on a HiSeq 2500 system with a 150-nt paired-end read protocol. Reads from the FASTQ files were processed by CutAdapt 1.15 to detect and trim adapter sequences. Processed sequences were aligned to the mouse genome assembly (GRCm38) using STAR and gene counts were extracted. Differential gene expression analysis was performed using a standard DESeq2 (v1.46.0) pipeline^80^ loaded in R^81^ (v4.5.0). Genes with log2(fold change) ≥ 0.58, and adjusted P-value ≤ 0.05 were considered significant. Gene Ontology(GO) terms of biological processes and pathway analysis were obtained using the DAVID functional annotation tool^82,83^, and dotplots for top enriched (P-value ranked) GO terms were constructed using clusterProfiler package (v4.14.4). Unsupervised k-means clustering and heatmap visualization was carried out using the ComplexHeatmap package^84^ using z-score normalized gene counts. For comparison with T2D datasets, human orthologues of mouse RNASeq data were generated using the Ensembl biomaRt webtool^85^. List of differentially expressed genes for Gpt2^βKO^ vs Gpt2^f/f^ are provided in Supplementary Tables 1 (control, 11mM glucose) and 2 (GLT).
Western Blot Analysis
HEK293T cells and islets were collected and washed twice in PBS at 4°C, pelleted at 300–800g and resuspended in RIPA buffer (150 mM NaCl, 50 mM Tris-HCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) with added protease and phosphatase inhibitors (Millipore Sigma, 539134 and 524625–1SET), and sonicated at highest power for 4–6 30s sonication: cooling cycles (Bioruptor Plus, Diagenode). Supernatants were collected after 20 minutes of centrifugation at 13000g, 4°C, and protein was quantified (BCA assay kit; Thermo Fisher, PI23235). Six-30 μg of protein lysates was mixed with sample loading buffer (Invitrogen, NP0007) and proteins were separated by 10 or 12% denaturing SDS-PAGE gel electrophoresis, followed by transfer to nitrocellulose membranes and blocking in 1X PBST + 5% blotting-grade blocker (Biorad, 1706404). Membranes were incubated with primary antisera overnight at 4°C, and HRP conjugated secondary antibody the next day for 2 hours at room temperature. After incubation with HRP substrate (Millipore, WBLUR0100), blots were visualized on a Chemi-doc Touch Imaging system (Bio-Rad) and densitometric quantification was performed using Fiji^86^. Details for primary and secondary antisera are provided in Supplementary Table 3.
Statistics
Values are reported as mean ±SE. To determine statistical significance between two groups, unpaired student’s t-test was used. One-way or two-way ANOVA with Tukey’s post hoc correction was used when comparing among multiple groups. P-value ≤ 0.05 was considered the threshold for considering significance. For performing hypergeometric test, the phyper function in R studio (v2024.09.0) was used and datasets showing FDR ≤ 0.05 were considered to have significant overlap. R studio (v2024.09.0) was used to create dot-plots and heatmaps. For all other statistics and graph plotting, GraphPad Prism v10 was used.
Supplementary Files
This is a list of supplementary files associated with this preprint. Click to download.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Zunz E. & Barre J. L. Contributions A L’Étude des Variations Physiologiques De La Sécrétion Interne Du Pancréas. Arch. Int. Physiol. (1929) doi:10.3109/13813452909145169. · doi ↗
- 2ELRICKH., STIMMLERL., HLADC. J.JR. & ARAIY. Plasma Insulin Response to Oral and Intravenous Glucose Administration 1. J. Clin. Endocrinol. Metab. 24, 1076–1082 (1964).14228531 10.1210/jcem-24-10-1076 · doi ↗ · pubmed ↗
- 3Mcintyre N., Holdsworth C. D. & Turner D. S. NEW INTERPRETATION OF ORAL GLUCOSE TOLERANCE. The Lancet 284, 20–21 (1964).10.1016/s 0140-6736(64)90011-x 14149200 · doi ↗ · pubmed ↗
- 4Nauck M., Stöckmann F., Ebert R. & Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 29, 46–52 (1986).3514343 10.1007/BF 02427280 · doi ↗ · pubmed ↗
- 5Drucker D. J. & Nauck M. A. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. The Lancet 368, 1696–1705 (2006).10.1016/S 0140-6736(06)69705-517098089 · doi ↗ · pubmed ↗
- 6Widenmaier S. B., Ao Z., Kim S.-J., Warnock G. & Mc Intosh C. H. S. Suppression of p 38 MAPK and JNK via Akt-mediated inhibition of apoptosis signal-regulating kinase 1 constitutes a core component of the beta-cell pro-survival effects of glucose-dependent insulinotropic polypeptide. J. Biol. Chem. 284, 30372–30382 (2009).19748889 10.1074/jbc.M 109.060178 PMC 2781592 · doi ↗ · pubmed ↗
- 7Yabe D. & Seino Y. Two incretin hormones GLP-1 and GIP: comparison of their actions in insulin secretion and β cell preservation. Prog. Biophys. Mol. Biol. 107, 248–256 (2011).21820006 10.1016/j.pbiomolbio.2011.07.010 · doi ↗ · pubmed ↗
- 8Dalle S., Burcelin R. & Gourdy P. Specific actions of GLP-1 receptor agonists and DPP 4 inhibitors for the treatment of pancreatic β-cell impairments in type 2 diabetes. Cell. Signal. 25, 570–579 (2013).23159576 10.1016/j.cellsig.2012.11.009 · doi ↗ · pubmed ↗