Synergistic solvent-catalyst paradigm for sustainable aerobic allylic C–H functionalization
Rui Wang, Long Zhang, Sanzhong Luo

TL;DR
A new sustainable method for allylic C–H functionalization uses a synergistic Pd/hydroquinone system with ethanol/water under mild conditions, achieving high efficiency and stereocontrol.
Contribution
The study introduces a synergistic solvent-catalyst system for sustainable allylic C–H functionalization with exceptional efficiency and pharmaceutical relevance.
Findings
A Pd/hydroquinone system enables aerobic allylic C–H functionalization at room temperature with high turnover frequency.
The method achieves over 90 products with up to 96% yield and 93% ee for quaternary stereocenters.
The platform integrates green solvent engineering and cooperative catalysis for scalable pharmaceutical synthesis.
Abstract
Achieving sustainable catalytic transformations requires synergistic optimization of solvent systems, catalytic motifs and energy inputs. Herein, we report a synergistic Pd/hydroquinone catalytic system that enables aerobic allylic C–H functions under ambient conditions (room temperature to 50°C, air) with high turnover frequency (TOF), using ethanol/water as a green medium. This strategy achieves unparalleled synthetic efficiency and demonstrates remarkable versatility across two pivotal transformations (alkylation and amination) involving over 90 products (up to 96% yield). It also delivers exceptional stereocontrol (up to 93% ee for quaternary stereocenters) and enables advanced allylic transformations within a green framework through additional synergistic catalysis. By integrating solvent engineering with cooperative catalysis, we have developed a scalable platform for the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
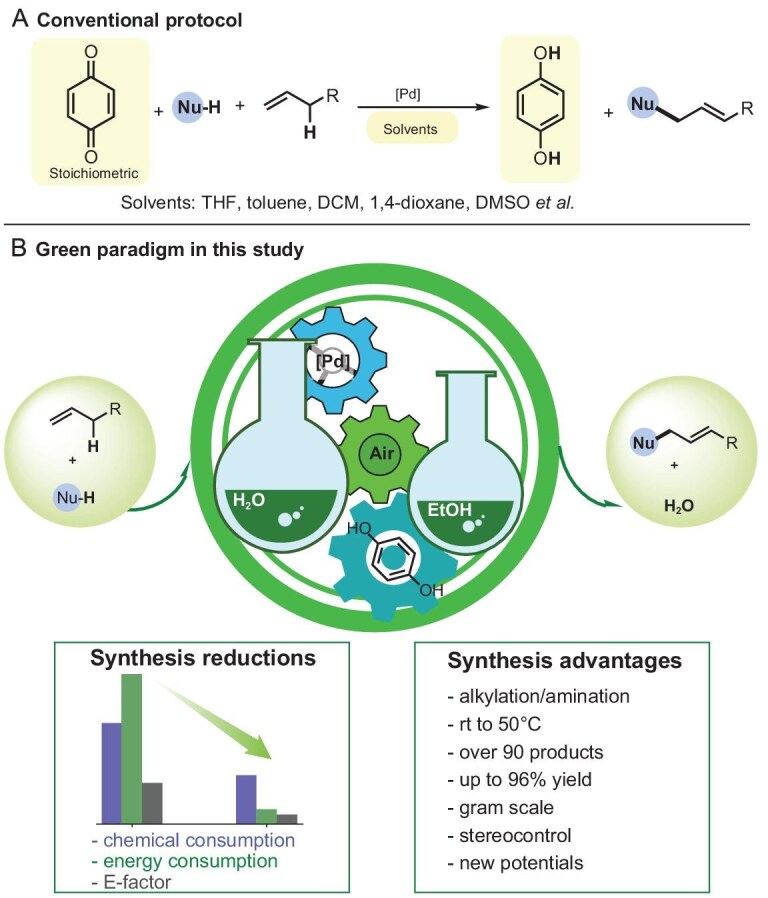 Figure 1
Figure 1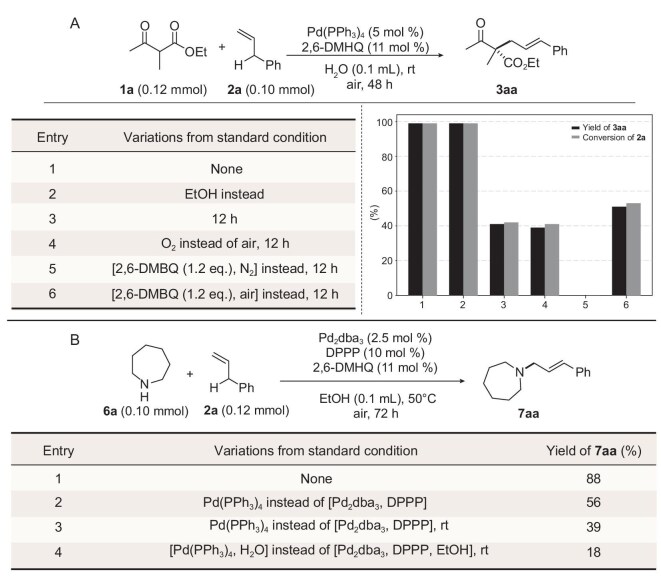 Figure 2
Figure 2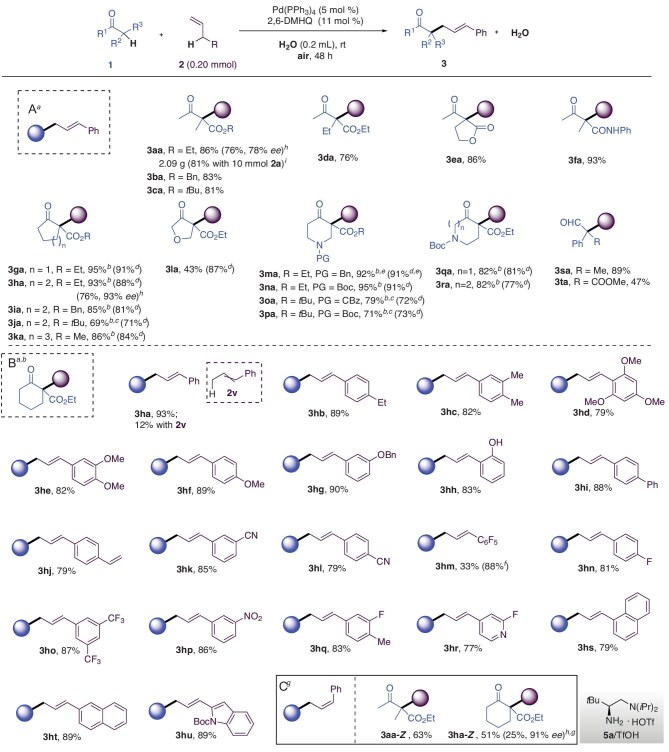 Figure 3
Figure 3 Figure 4
Figure 4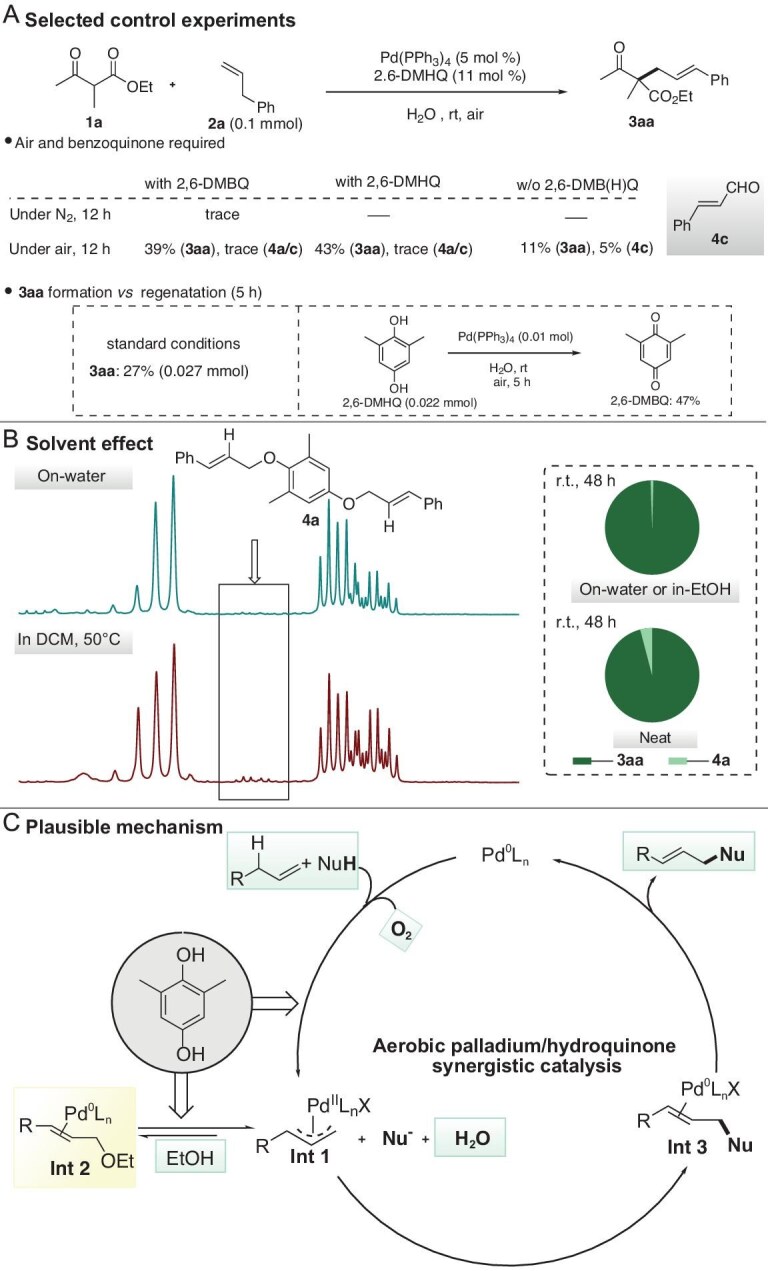 Figure 5
Figure 5- —National Natural Science Foundation of China10.13039/501100001809
- —National Key R&D Program of China10.13039/501100012166
- —Haihe Laboratory of Sustainable Chemical Transformations
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCatalytic C–H Functionalization Methods · Asymmetric Hydrogenation and Catalysis · Synthesis and Catalytic Reactions
INTRODUCTION
In the global pursuit of sustainable and environmentally benign chemical processes, palladium-catalyzed allylic C–H functionalization has emerged as an efficacious transformative strategy in synthetic chemistry, enabling direct construction of C–C and C–X bonds with inherent atom economy [1–11]. While palladium-catalyzed allylic chemistry has seen remarkable advances, its industrial adoption remains hampered by reliance on stoichiometric benzoquinone oxidants and hazardous solvents—critical barriers to achieving carbon-neutral chemical production as outlined in China's 14th Five-Year Plan [12] (Fig. 1A). Against this backdrop, the development of green and efficient catalytic systems is not only a scientific imperative but also a strategic necessity for the fine chemical industry [13–16].
Pioneering studies by Stahl et al. [17–21], Yu et al. [22–24], Báckvall et al. [25–27] and others [28–35] have established the viability of aerobic palladium catalysis. Building on these advances, our previous work demonstrated that molecular oxygen (O_2_) from ambient air serves as an ideal green oxidant for aerobic allylic alkylation, leveraging its sustainability advantages—abundance, low cost and environmentally benign byproduct (H_2_O). However, critical challenges persist, including low reaction rates and reliance on hazardous solvents [36], which hinder broader industrial adoption. Moreover, a generalizable methodology for aerobic allylic C–H activation remains elusive. For instance, while recent breakthroughs by White et al. [37], Jiang et al. [38,39] and Gong et al. [40] have advanced allylic amination chemistry, these Pd-catalyzed platforms still necessitate stoichiometric benzoquinone oxidants. Our current research represents an effective approach to overcoming these limitations by integrating solvent engineering with cooperative catalysis.
Water and ethanol, as universal, safe and sustainable solvents, are often overlooked in organic chemistry due to solubility limitations and sensitivity of reactants (hydrolysis or alcoholysis) [41–43]. Additionally, the extra reducing and nucleophilic properties of ethanol make it a cautious choice and rarely examined as a solvent in organic reactions [43–46]. Our findings, which began with the observation that undried dichloromethane (DCM) improved reaction rates in aerobic allylic C–H activation [36], led us to investigate the use of water (the most ideal green solvent) [13,15]. While ethanol, compliant with the CHEM21 Solvent Guide [47] and Chinese standards HG/T 5970–2021 and GB 31604.60–2024, serves as a highly viable alternative solvent for enhancing the conversion of specific substrates. Our work leverages these green solvents to achieve broad-spectrum allylic C–H activation, including alkylation, amination and dehydrogenation, with significant benefits for green chemistry. This approach further enables stereoselectivity in alkylation, direct formation of a Z-type allylic compound [48–50] and sequential bis-allylic amination of primary amines, offering a sustainable blueprint for fine chemical synthesis. Compared with conventional oxidative allylic C–H functionalizations (e.g. amination and asymmetric alkylation) that yield partially overlapping products, our method significantly reduces the E-factor (environmental factor) and reaction energy consumption (Figs S17 and S18). Preliminary mechanistic studies reveal that the green solvents, in conjunction with air, play a crucial role in enhancing overall catalytic efficiency. Specifically, the green solvents help maintain the activity of hydroquinone (or quinone, the core catalyst for aerobic reactions) and may also enhance the efficiency of palladium catalysis through different pathways to the target products, with this effect being particularly notable in ethanol. This research provides a sustainable and efficient strategy for allylic transformations, emphasizing the importance of both synthetic innovation and green chemistry principles (Fig. 1B).
RESULTS AND DISCUSSION
Condition optimizations
We have systematically optimized reaction conditions for allylic C–H alkylation and amination, using ethanol or water as green solvents under ambient air conditions. For the alkylation reaction, employing Pd(PPh_3_)4 (5 mol%) and 2,6-DMHQ (11 mol%) on water or in ethanol at room temperature (rt) achieved a remarkable yield of >99% for 3aa within 48 hours (Fig. 2A, entries 1 and 2). This significantly outperformed reactions conducted in a variety of commonly used organic solvents, which showed only trace amounts of product at rt (for detailed solvent and condition screening, including benzoquinones, palladium sources and ligands, see Figs S1–S4). We found that the yield of the target product 3aa was comparable in air and oxygen under our operational conditions (Fig. 2A, entries 3 and 4; for the detailed procedure, see Supplementary data). Consequently, air was chosen as the more versatile oxygen source for substrate investigation, while oxygen was employed for gram-scale reactions to facilitate operational ease. Additionally, stoichiometric utilization of 2,6-DMBQ revealed that oxidative allylic C–H alkylation was ineffective at rt under N_2_, while 3aa was achieved under ambient air conditions (Fig. 2A, entries 5 and 6).
Synergistic solvent-catalyst paradigm for aerobic allylic C–H functionalization.
Control experiments. (A) Alkylation: 1a (0.12 mmol), 2a (0.10 mmol), Pd(PPh3)4 (5 mol %), 2,6-DMHQ (11 mol %), H2O (0.1 mL), rt, 48 h, air. (B) Amination: 6a (0.10 mmol), 2a (0.12 mmol), Pd2dba3 (2.5 mol %), 2,6-DMHQ (11 mol %), EtOH (0.1 mL), 50°C, 72 h, air. The yield was determined by 1H NMR with CH2Br2 as internal standard.
For the amination reaction, using Pd₂(dba)₃ in combination with the DPPP ligand in ethanol at 50°C delivered the optimal yields (Fig. 2B, entry 1, 88% for compound 7aa). This protocol effectively overcame the reaction rate limitations observed with the alkylation conditions used in migration processes (Fig. 2B, entries 3 and 4) and the heating process in EtOH (Fig. 2B, entry 2). Detailed screening of various conditions can be found in Figs S5–S8.
Our ‘green solvent–air’ system also enabled other various transformations, including fine stereocontrol for quaternary centers, sequential dehydrogenation to form conjugated dienes, etherification and more functions, all with high efficiency (Figs S9–S12).
Scope of alkylation
The protocol demonstrated broad substrate compatibility (Fig. 3). β-Ketocarbonyl compounds (esters, amides, lactones) and α-branched aldehydes reacted efficiently with allyl arenes to form all-carbon quaternary centers (3aa–3ta, 76%–95% yield). For instance, the incremental addition of 2,6-DMHQ to bulky substrates (e.g. t-butyl esters forming 3ja, 3oa and 3pa) was necessary to maintain reactivity on water, while the reaction proceeded robustly in ethanol. The synthesis of 3la was more efficient in ethanol despite complete substrate conversion on water.
Alkylation scope: (A) with respect to the β-ketocarbonyl and α-branched aldehydes substrates; (B) with respect to the allyl arenes substrates; (C) Z-retentive allylic substitution. aReaction conditions: 1 (0.24 mmol), 2 (0.20 mmol), Pd(PPh3)4 (5 mol %), 2,6-DMHQ (11 mol %), H2O (0.4 mL), rt, 48 h, air; isolated yield. bPd(PPh3)4 (8 mol %), 2,6-DMHQ (18 mol %) used; isolated yield. cAnother 18 mol % 2,6-DMHQ added after 24 h. dEtOH (0.4 mL) instead of H2O as solvent. e1 (0.20 mmol), 2a (0.24 mmol) used. fAt 50°C. gAdditional Ir(ppy)3 (2.5 mol %) added, 30 W blue LED. hReaction conditions: 1 (0.15 mmol), 2a (0.10 mmol), 5a/TfOH (12 mol %), Pd(PPh3)4 (10 mol %), 2,6-DMHQ (22 mol %), H2O (0.15 mL), rt, 48 h, air; isolated yield; the ee was determined by HPLC. iGram scale: 1a (12 mmol), 2a (10 mmol), Pd(PPh3)4 (5 mol %), 2,6-DMHQ (11 mol %), EtOH (10 mL), rt, 48 h, O2; isolated yield.
Stereocontrol was achieved via a chiral primary amine 5a/TfOH co-catalytic system, yielding 3aa in 76% yield with 78% ee and 3ha in 76% yield with 93% ee (H_2_O was crucial for maintaining enantioselectivity, outperforming EtOH, see Fig. S9).
The compatibility of various allyl arenes 2 with ethyl 2-cyclohexanone-1-carboxylate 1h was assessed under on-water conditions using the Pd-hydroquinone synergistic catalyst (Fig. 3B). Allyl arenes with electron-donating (e.g. methyl, ethyl, methoxy) to strong electron-withdrawing substitutions (e.g. –CN, –F, –CF_3_, –NO_2_) showed good to excellent reactivity (forming 3hb–3hl and 3hn–3hq, 79%–93% yield). Allyl pentafluorobenzene gave 3hm in good yield at 50°C. 4-Allyl-2-fluoropyridine without protection on pyridine nitrogen afforded product 3hr in acceptable 77% yield. 2-Allylnaphthalene served as a good substrate to offer 3ht in 89% yield, while 1-allylnaphthalene with higher steric resistance showed limited reactivity to offer 3hs. The reaction with an indole-derived substrate also proceeded well to offer 3hu in 89% yield. Internal alkenes such as 1-propenylbenzene 2v was examined, showing rather poor reactivity.
When irradiated by a 30 W blue LED with Ir(ppy)3 (2.5 mol %), the reaction led to the formation of Z-retentive allylic substitution 3aa-Z and 3ha-Z (Fig. 3C), albeit with relatively lower yields due to the side reaction of 2,6-DMHQ. While without irradiation, it turned back to E-retentive allylic substitution 3aa and 3ha mediated by the palladium catalyst. Under photo catalysis with the synergistic Ir(ppy)3 (2.5 mol %), the asymmetric catalysis with our chiral primary amine 5a/TfOH co-catalytic system produced Z-retentive allylic substitution 3ha-Z in 25% yield, still maintaining high enantioselectivity with 91% ee.
To evaluate the practicality of the method, a gram-scale alkylation to construct all-carbon quaternary product 3aa was performed under optimized conditions in ethanol with O_2_ as the sole oxidant, yielding the desired allylic products in 81% yield.
Scope of amination
Under optimized amination conditions, the substrate scope was examined (Fig. 4). A wide range of amines, including aliphatic primary amines, secondary amines and anilines, underwent the reaction efficiently (Fig. 4A). Cyclic alkyl amines provided products 7aa–7ca in fine to good yields. Chain secondary alkyl amines afforded 7da–7ga in considerable to excellent yields. Oxocyclic amines yielded 7ha and 7ja in >90% yield, while thiomorpholine gave 7ia in a moderate yield. Notably, morpholine exhibited special reactivity under optimized conditions. Secondary amines such as piperazine derivatives could be converted to the desired products 7ka–7oa in high yields, mainly with augmenting of the Pd-hydroquinone synergistic catalyst. The reduced activity of piperazine derivatives may be due to the increased alkalinity of the substrates.
Amination scope: (A) with respect to the amine substrates; (B) with respect to the allylic substrates; (C) bis-allylic amination; (D) gram-scale synthesis. aReaction conditions: 6 (0.20 mmol), 2 (0.24 mmol), Pd2(dba)3 (2.5 mol %), DPPP (10 mol %), 2,6-DMHQ (11 mol %), EtOH (0.2 mL), 50°C, 84 h, air; isolated yield. b72 h. cPd(PPh3)4 (5 mol %), rt, 72 h. d6k (0.10 mmol), 2 (0.24 mmol), Pd2(dba)3 (10 mol %), DPPP (40 mol %), 2,6-DMHQ (44 mol %), EtOH (0.2 mL), 50°C, 84 h, air; ePd2(dba)3 (5 mol %), DPPP (20 mol %), 2,6-DMHQ (22 mol %) used. f6’ (0.24 mmol), 2a (0.20 mmol), Pd2(dba)3 (2.5 mol %), DPPP (10 mol %), 2,6-DMHQ (11 mol %), EtOH (0.2 mL), 50°C, 72 h, air. g6’ (0.24 mmol), 2a (0.20 mmol), Pd(PPh3)4 (5 mol %), 2,6-DMHQ (11 mol %), EtOH (0.2 mL), 50°C, 72 h, air; the yield was determined by 1H NMR with CH2Br2.h6’ (0.20 mmol), 2a (0.50 mmol), Pd2(dba)3 (2.5 mol %), DPPP (10 mol %), 2,6-DMHQ (11 mol %), EtOH (0.2 mL), 50°C, 96 h, air. iPd2(dba)3 (5 mol %), DPPP (20 mol %), 2,6-DMHQ (22 mol %), EtOH (0.4 mL) used. j2a (5 equiv.), Pd(PPh3)4 (5 mol %), 2,6-DMHQ (11 mol %), H2O (0.1 mL), O2 used; rt. kWith 2v. l96 h. m6d’ (0.20 mmol), 2a (0.20 mmol), 2m or 2x (0.50 mmol), Pd(PPh3)4 (5 mol %), 2,6-DMHQ (11 mol %), EtOH (0.2 mL), rt, 48 h, air; then 50°C, 72 h, O2. nGram scale: 6 (2.5 mmol), 2 (5 mmol), Pd2(dba)3 (2.5 mol %), DPPP (10 mol %), 2,6-DMHQ (11 mol %), EtOH (2.0 mL), 50°C, 84 h, O2; isolated yield.
Secondary anilines were effectively utilized under ‘room-temperature-on-water’ amination conditions to provide good results, such as 8aa. Aromatic primary amines could offer mono- and di-allylation products in fine to excellent yields with exclusive chemoselectivity under separate optimized conditions. With aromatic primary amines 6 (0.24 mmol) bearing different electronic/site substituents and allyl benzene 2a (0.20 mmol), mono-allylation products 8ba-mono–8ja-mono were obtained in up to 93% yield within 72 hours. The yield of 8ha-mono was low, likely due to the poor solubility of the amine substrate. The bromination substrate intimated derivative potentiality was transformed into 8ga-mono in fine yield; 8ka-mono was synthesized on water in fine yield, but the reaction did not proceed in ethanol, likely due to the lower nucleophilicity and concentration of pentafluoroaniline. With aromatic primary amines 6 (0.20 mmol) bearing electron-donating substituents and allyl benzene 2a (0.50 mmol), di-allylation products 8-di were obtained in up to 91% yield within 96 hours.
Alkyl primary amines could also be converted to desired products in good yield using a similar protocol, such as 9aa. Considerable tri-allylation 9ba was achieved with a tyrosine derivative on water under an oxygen atmosphere.
The compatibility of various allylic substrates 2 with morpholine 1h was also evaluated (Fig. 4B). Allyl arenes bearing electron-donating substituents provided 7hd/7he/7hg in >80% yield. A free hydroxyl substituted 2-allylphenol afforded 7hh in 78% yield. Olefin-substituted allyl arenes also furnished desired 7hi in 87% yield with the other double bond remaining intact. Pentafluoroallylbenzene bearing strong electron-withdrawing substituents showed relatively low reactivity but still provided 7hm in 73% yield within 96 hours. 2-Allylnaphthalene served as a good substrate to offer 7ht in 85% yield. Alkyl alkene 4-phenyl-1-butene provided 7hw in 37% yield, while 1-propenylbenzene 2v offered 7ha in 27% yield.
Based on the different reactivity of allylic substrates, a sequential bis-allylic C–H amination with primary amines was also achieved using the developed synergistic catalysis; 8dam and 8dax were obtained in satisfactory yields under one-pot conditions (8da–di <20%, the sum of all other allylic products <10%, Fig. 4C, Fig. S12).
Gram-scale amination to form FDA-certified biologically active allylic amines 7pe (inhibitor of ABA biosynthesis), 7ga (antifungal agent), 7na (calcium antagonist) and 7oa (calcium antagonist) was performed under optimized conditions in ethanol with O_2_ as the sole oxidant, yielding the desired products in good to excellent yields.
Mechanism studies
We explored the mechanism underlying our ‘green solvent-empowered’ aerobic allylic C–H functionalization facilitated by synergistic Pd-hydroquinone catalysis. Control experiments revealed that the alkylation reaction was completely suppressed at rt when either (hydro)quinone or oxygen was absent (Fig. 2A, entries 5 and 6, and Fig. 5A; Figs S4, S13 and S14; Fig. S7 for amination). Hydroquinone (2,6-DMHQ) demonstrated similar and even slightly better performance when compared with its quinone (2,6-DMBQ) precursor (Fig. 5A, details in Figs S4 and S7). In a separate experiment, it was found that the regeneration of 2,6-DMBQ from its hydroquinone counterpart 2,6-DMHQ was much slower than the formation of 3aa under identical aerobic Pd catalysis (Fig. 5A). These critical findings collectively suggest that the current catalytic system operates through a mechanism distinctive from both the conventional quinone–regeneration-driven C–H activation processes [17,25] and the direct quinone mediated C–H activation process as previously reported by Gong and White [15,34,51–54].
Mechanism studies. (A) control experiments; (B) solvent effects; (C) proposed catalytic cycle.
A further increase in the loading of hydroquinone 2,6-DMHQ led to only minor improvement on activity along with the accumulation of side-adducts such as 4a (Fig. S4, Appendix B). Under aerobic conditions, minor yet notable background reactivity was observed in the absence of quinone/hydroquinone cocatalyts (Fig. 5A). Thereby we arrived at a key conclusion: (hydro)quinone likely does not follow typical regeneration pathways but instead synergizes with air to facilitate allylic C–H functionalization during the palladium cycle (Fig. 5A). Recent studies have shown that stoichiometric reductive species combined with O_2_ can boost the oxygen reduction reaction (ORR) and the efficiency of cascade oxidation of substrates [55], which aligns with our observations.
In conventional organic solvents such as DCM, the synergistic Pd/(hydro)quinone showed rather low and even no activity (Fig. S3). In these instances, hydroquinone loses its catalytic activity upon etherification via electrophilic attack on the π-allylpalladium intermediate, leading to etherification byproducts as experimentally observed [45]. In stark contrast, reactions conducted in H_2_O (except those involving bulky nucleophiles requiring additional 2,6-DMHQ) show no detectable etherification byproducts 4a by NMR analysis (Fig. 5B). Although ethanol-mediated reactions consistently generate trace etherification products, arising from ethanol as nucleophile, these do not impede the thermodynamically favored alkylation/amination ratio (Fig. S3, entry 15), which is consistent with the reversible allylic etherification processes reported in recent oxidative allylic functionalization studies [45]. On the other hand, the reaction under neat conditions, though equally fast, was unavoidably accompanied with a considerable amount of side products such as 4a (Fig. 5B). In addition, the solid nature of specific substrates also limited the applicability of the neat conditions. These results highlight the beneficial features of water and ethanol as green solvents.
We attribute this solvent-driven enhancement to two key mechanisms: (1) water or ethanol can largely suppress the nucleophilic attack of 2,6-DMHQ on π-allylpalladium species through solvation, thereby maintaining the activity of the (hydro)quinone co-catalyst (Fig. 5C); (2) the protic and polar nature may facilitate the palladium catalytic cycles. In ethanol, these paths include oxidative C–H activation (Fig. 5C, formation of Int1) and reductive functionalization of etherification byproducts (Fig. 5C, from Int2 to Int1). In the control experiment in the absence of nucleophiles, effective etherification was observed and the obtained allylic ethyl ether underwent smooth alkylation when nucleophiles were supplied (Fig. S14). With water, the on-water nature may facilitate the overall process by hydrophobic concentration effects as well as the known interphase O–H effect.
In summary, we have preliminarily proposed a plausible mechanism for the reaction (Fig. 5C), involving synergistic palladium/hydroquinone catalysis, solvent and air working together to achieve the target product.
CONCLUSION
By integrating solvent engineering with cooperative Pd/hydroquinone catalysis, we have established a sustainable platform for allylic C–H functionalization that achieves unparalleled efficiency while eliminating wasteful oxidants and toxic reagents. This system aligns with the general trends in sustainable chemistry and directly addresses the strategic goals of China's green chemistry agenda. Mechanistic studies have revealed the key roles of palladium activity in a novel parallel-mechanism paradigm, where we have utilized green solvents to maximize the turnover number (TON) and turnover frequency (TOF) for specific allylic product synthesis and highlight the potential for further innovation. This work bridges molecular innovation with industrial decarbonization and provides a blueprint for green pharmaceutical synthesis, aligning with current sustainable chemistry efforts.
METHODS
Full experimental details and characterization of the compounds are given in the Supplementary data.
Supplementary Material
nwaf196_Supplementary_File
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Trost MB, Fullerton JT. New synthetic reactions. Allylic alkylation. J Am Chem Soc 1973; 95: 292–4.10.1021/ja 00782 a 080 · doi ↗
- 2Li Z, Li C. Catalytic allylic alkylation via the cross-dehydrogenative-coupling reaction between allylic sp 3 C–H and methylenic sp 3 C–H bonds. J Am Chem Soc 2006; 128: 56–7.10.1021/ja 056541 b 16390119 · doi ↗ · pubmed ↗
- 3Lin S, Song C, Cai G et al. Intra/intermolecular direct allylic alkylation via Pd(II)-catalyzed allylic C–H activation. J Am Chem Soc 2008; 130: 12901–3.10.1021/ja 803452 p 18778061 · doi ↗ · pubmed ↗
- 4Li C . Cross-dehydrogenative coupling (CDC): exploring C–C bond formations beyond functional group transformations. Acc Chem Res 2009; 42: 335–44.10.1021/ar 800164 n 19220064 · doi ↗ · pubmed ↗
- 5Liu G, Wu Y. Palladium-catalyzed allylic C–H bond functionalization of olefins. In: Yu JQ, Shi Z (eds). C-H Activation. Top Curr Chem 2009; 292: 195–209.10.1007/128_2009_1621500407 · doi ↗ · pubmed ↗
- 6Qi X, Chen P, Liu G. Avances and challenges in palladium-catalyzed intermolecular selective allylic C–H functionalization of alkenes. Sci China Chem 2015; 58: 1249–51.10.1007/s 11426-015-5387-9 · doi ↗
- 7Liu C, Zhang H, Shi W et al. Bond formations between two nucleophiles: transition metal catalyzed oxidative cross-coupling reactions. Chem Rev 2011; 111: 1780–824.10.1021/cr 100379 j 21344855 · doi ↗ · pubmed ↗
- 8Engelin CJ, Fristrup P. Palladium catalyzed allylic C–H alkylation: a mechanistic perspective. Molecules 2011; 16: 951–69.10.3390/molecules 1601095121258300 PMC 6264533 · doi ↗ · pubmed ↗