Imine-Oxazoline (ImOx): A C 1‑Symmetric N,N‑Bidentate Ligand for Asymmetric Catalysis
Elliot S. Silk, Haozhe Zhu, Alexander G. Shtukenberg, Tianning Diao

TL;DR
This paper introduces a new chiral ligand, ImOx, designed to improve asymmetric catalysis by enhancing enantioselectivity and enabling new reaction pathways.
Contribution
The novel ImOx ligand combines features of α-diimine and pyridine oxazoline ligands with tunable steric effects for asymmetric catalysis.
Findings
ImOx improves enantioselectivity in palladium-catalyzed conjugate addition reactions.
The steric bulk of ImOx necessitates a cationic pathway for alkene insertion.
ImOx shows stronger trans-influence and versatile redox activity compared to PyOx.
Abstract
Asymmetric catalysis relies on the design of chiral ligands, but the variety of nitrogen-based ligands remains limited. To address this gap, we have developed a class of C 1-symmetric N,N-bidentate ligands, imine-oxazoline (ImOx), derived from amino acids through a four-step synthesis. ImOx features an imine moiety conjugated with a chiral oxazoline ring as a hybrid of α-diimine (ADI) and pyridine oxazoline (PyOx) ligands. Its low symmetry allows for independent optimization at both coordination sites. ImOx improves the enantioselectivity of palladium-catalyzed conjugate addition reactions, demonstrating a strong correlation between ee and the steric effects on both the imine and oxazoline sites. Studies on well-defined organopalladium intermediates reveal that the steric bulk of ImOx necessitates a cationic pathway to promote alkene insertion. Structural characterization of ImOx…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1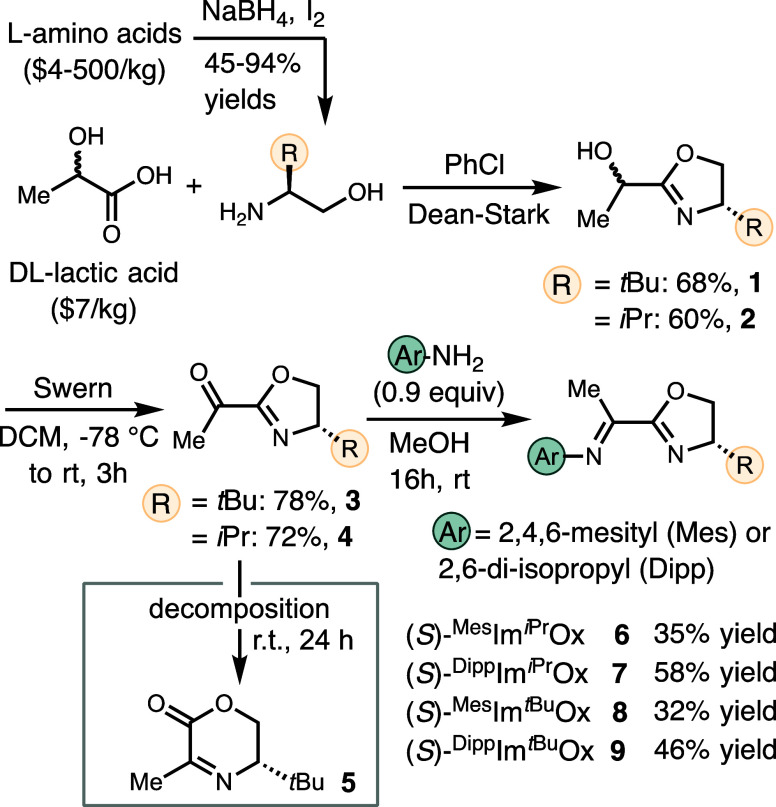 2
2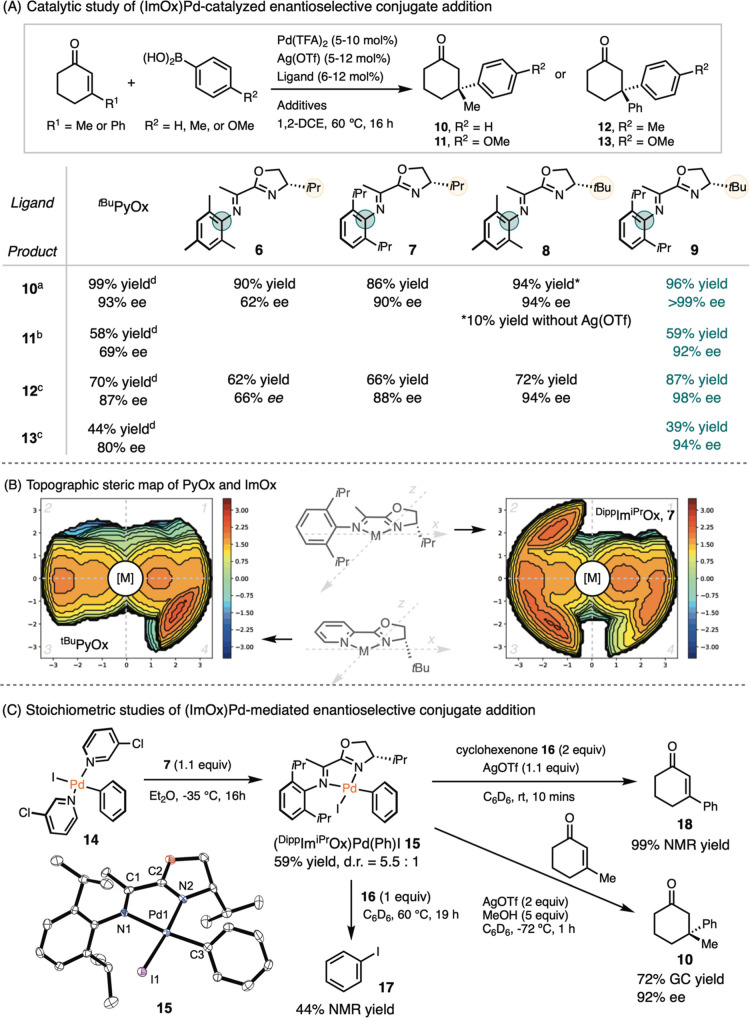 3
3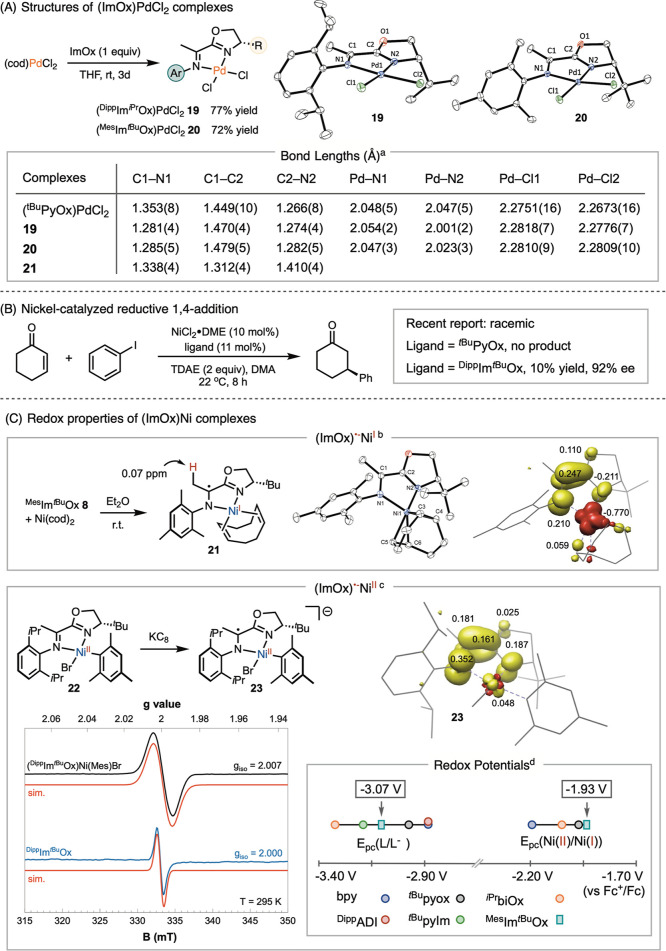 4
4- —National Institute of General Medical Sciences10.13039/100000057
- —Division of Graduate Education10.13039/100000082
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAsymmetric Hydrogenation and Catalysis · Asymmetric Synthesis and Catalysis · Chemical Synthesis and Analysis
Introduction
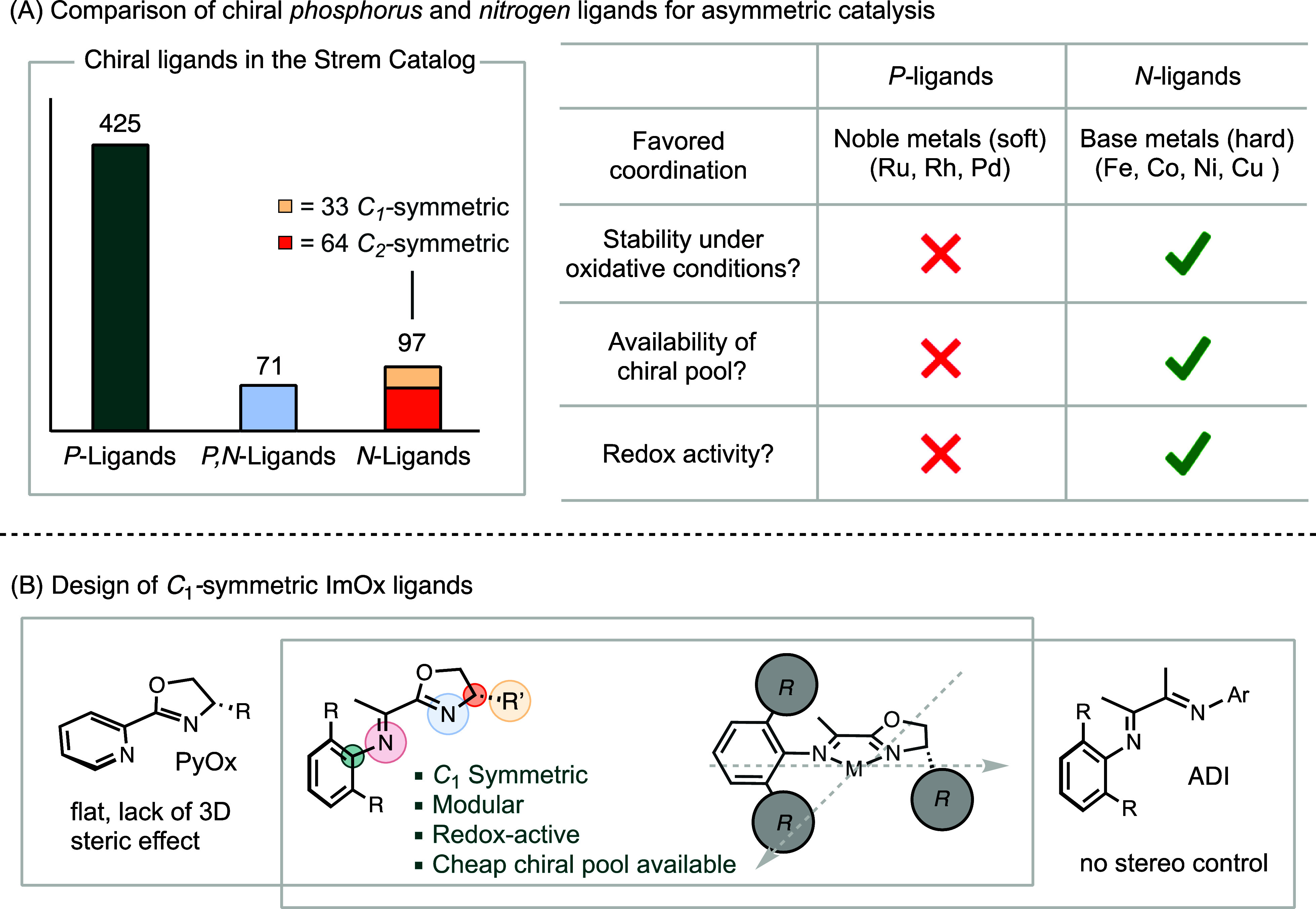
The stereochemical configuration of a drug can significantly affect its potency, specificity, and pharmacokinetics. Currently, over 90% of chiral molecules are synthesized using stoichiometric methods, including derivatization from chiral pools, resolution, and chiral transfer from auxiliaries and reagents.? Asymmetric catalysis offers a sustainable, atom-economic, and efficient alternative for introducing chirality in pharmaceutical targets, though its scope and enantioselectivity are largely dependent on the availability of chiral ligands. ?,?
While a large library of phosphorus ligands is commercially available, driven by the success of asymmetric hydrogenation,? chiral nitrogen ligands remain underdeveloped.? Among commercially available chiral ligands, there are more than four times as many chiral phosphorus ligands compared to nitrogen ligands (SchemeA). Recently, nitrogen ligands have significantly advanced base-metal catalysis. ?−? ? ? These ligands coordinate favorably to 3d metals according to the hard–soft acid–base (HSAB) principle and exhibit better stability under oxidative conditions. Moreover, pyridine and Schiff base derivatives can engage in redox activity, stabilizing metalloradical intermediates.? The chirality of nitrogen ligands often leverages the naturally abundant chiral pool of amino acids,? which is more straightforward compared to the methods required for synthesizing phosphorus ligands through resolution or asymmetric coupling. ?,?
Motivation and Design Principles of Imine-Oxazoline (ImOx) Ligands for Asymmetric Catalysis
The majority of commercial N-based bidentate chiral ligands are C 2-symmetric, such as bis-oxazoline (box) analogues. ?,? A lower symmetry bidentate ligand with two different coordinating sites allows for independent optimization of individual coordination sites. ?,? This consideration has led to the design of C 1-symmetric pyridine-oxazoline (PyOx). ?,? Additionally, α-diimine (ADI) ligands offer versatile modularity for tuning steric and electronic effects. ?,? Here, we report the development of a C 1-symmetric bidentate imine-oxazoline (ImOx) ligand by hybridizing the imine moiety of ADI and the chiral oxazoline component of PyOx (SchemeB). A related ImOx variant has been described before, ?−? ? featuring a phenyl group in the backbone. While the phenyl group is necessary for the previously developed synthetic routes, its steric hindrance limits potential applications in asymmetric catalysis.? In this work, we have developed an entirely new synthetic route that enables access to the catalytically relevant ImOx variant with a methyl backbone. This highly modular ligand leverages the natural chiral pool of amino acids and is accessible in four steps. With compelling electronic and steric properties, ImOx has proven effective in facilitating enantioselective conjugate additions.?
Methods
We initiated our study by developing the synthesis of ImOx scaffolds, focusing on promoting accessibility and minimizing cost. The availability of amino acids as a chiral pool and dl-lactic acid? as abundant, naturally occurring building blocks prompted us to explore a route starting with the condensation of amino alcohols with dl-lactic acid to afford α-methyl oxazolyl alcohols 1 and 2 (Scheme). ?,? The oxidation of 1 and 2 proved challenging, which might account for the lack of precedents for ImOx ligands. Dess–Martin periodinate, pyridinium chlorochromate, and aerobic ABNO conditions ?,? appeared incompatible with the oxazoline functionality, leading to substrate decomposition. Swern oxidation of 1 or 2 with (COCl)2 and DMSO gave ketones 3 or 4, respectively, in good yields. Initially, we tried to isolate and purify 3 or 4, but observed a spontaneous rearrangement of 3 to the six-membered ring 5.? Thus, we condensed 3 or 4 with anilines without intermediate purification to afford ImOx ligands: (S)-^Mes^Im^ iPr^Ox (6), (S)-^Dipp^Im^ iPr^Ox (7), (S)-^Mes^Im^ tBu^Ox (8), and (S)-^Dipp^Im^ tBu^Ox (9) (iPr = isopropyl, tBu = tert-butyl, Mes = 2,4,6-trimethylphenyl, Dipp = 2,6-diisopropylphenyl).
Synthesis of ImOx Ligands from Lactic Acid and Amino Acids
With a range of ImOx variants in hand, we explored their utility in asymmetric catalysis. Palladium-catalyzed conjugate addition to α,β-unsaturated enones has emerged as an effective strategy for constructing all-carbon quaternary centers. tBuPyOx has been reported to facilitate the synthesis of 10 with high yield and excellent ee, ?,? whereas its application to the synthesis of the diaryl derivative 11 resulted in modest yield and ee.? We examined the performance of ImOx in these two reactions in comparison to PyOx.
Initially, we replaced tBuPyOx with MesImtBuOx 8 in the original conditions for palladium-catalyzed asymmetric conjugate addition,? but observed 10 in only 10% yield, along with the formation of biphenyl and significant catalyst decomposition into palladium black (SchemeA). We attributed the catalyst decomposition to the formation of a Pd-bis-aryl intermediate, followed by reductive elimination to form Pd(0). To address this competing pathway, we added AgOTf as a Lewis acid and counterion abstractor to facilitate enone coordination and subsequent insertion. AgOTf is also capable of reoxidizing Pd(0) to Pd(II), potentially preventing Pd black formation.? Furthermore, we improved the yield by introducing five equivalents of H_2_O to promote transmetalation by generating the active boronate species.?
Application of ImOx to Palladium-Catalyzed Conjugate Addition to Form Quaternary Chiral Centers
Comparisons among ImOx ligands 6–9 indicated that bulkier substituents on both the aryl and oxazoline moieties led to increased yield and ee (SchemeA). The effect of substituents on the oxazoline ring appeared to be more significant than those on the imine. Notably, (S)-^Dipp^Im^ tBu^Ox 9 delivered a quantitative yield and perfect ee, with no minor enantiomer detected by HPLC (Figure S5). Applying (S)-^Dipp^Im^ tBu^Ox 9 to a more challenging substrate, such as para-methoxyphenyl boronic acid nucleophile, improved the ee of 11 from 69% as previously reported? to 92%.
Subsequently, we evaluated the application of ImOx to the more challenging conjugate addition of p-tolylboronic acid to 3-phenylcyclohexenone to afford 12 (SchemeA).? Compared to the initial conditions reported with PyOx,? the use of ImOx required the use of AgOTf as an additive to prevent catalyst decomposition and achieve high yields. Additionally, the reaction with ImOx benefited from the addition of 5 equiv of MeOH or water to facilitate transmetalation, along with Sc(OTf)3 as a Lewis acid to activate the enone, and 18-crown-6 as a phase transfer catalyst. Under these optimized conditions, we evaluated the effect of ImOx variants. The trend of ligand effect was similar to that observed in the formation of 10. Bulkier substituents on both the imine and oxazoline moieties improved the yield and ee. A comparison between (S)-^Mes^Im^ iPr^Ox 6 and (S)-^Dipp^Im^ iPr^Ox 7 revealed that replacing Mes with Dipp on the imine improved the ee from 66% to 88%. The substituent on oxazoline had an even more profound effect, increasing the ee from 66% to 94% when the iPr group of (S)-^Mes^Im^ iPr^Ox 6 was switched to a tBu group in (S)-^Mes^Im^ tBu^Ox 8. A high yield of 87% and an ee of 98% were obtained with (S)-^Dipp^Im^ tBu^Ox 9. Using (S)-^Dipp^Im^ tBu^Ox 9 with the more challenging para-methoxyphenyl boronic acid enhanced the ee of 13 from the previously reported 80% to 94%.
Control experiments using ^ tBu^PyOx under our optimized conditions afforded 10 in an 88% yield and 90% ee, and 12 in a 29% yield and 86% ee (cf. Supporting Information). These results suggest that the observed improvement in enantioselectivity arises from the ImOx ligand itself, rather than from changes in reaction conditions. Steric heat maps? shed light on the differences between ImOx and ^ tBu^PyOx (SchemeB). In the case of ^ tBu^PyOx, the tBu group on the oxazoline occupies quadrant four, leaving quadrants one, two, and three relatively open. In contrast, ImOx spans quadrants two, three, and four, thereby restricting the approach vectors of the substrate toward the metal catalyst.
While the mechanism of (PyOx)Pd-catalyzed conjugate addition has been well-understood,? we carried out organometallic studies of (ImOx)Pd for comparison. Ligand substitution of bis(3-chloropyridine)Pd^II^(Ph)I 14 ? with (S)-^Dipp^Im^ iPr^Ox 7 resulted in the formation of (^Dipp^Im^ iPr^Ox)Pd(Ph)I 15 as a mixture of diastereomers in a ratio of 5.5:1 (SchemeC). NOESY experiments established that the major diastereomer has the phenyl group trans to imine (Figure S45). Single-crystal X-ray analysis of 15 confirmed a square planar geometry.
When we subjected 15 to one equivalent of cyclohexenone 16 and heated the mixture to 60 °C overnight, we observed formation of iodobenzene 17 in 44% yield. In the presence of 1.1 equiv of AgOTf, (^Dipp^Im^ iPr^Ox)Pd(Ph)I 15 fully converted into 3-phenylcyclohexenone 18 via insertion followed by β-H elimination. When we replaced cyclohexenone 16 with 3-methylcyclohexenone in the presence of five equivalents MeOH as a proton source, (^Dipp^Im^ iPr^Ox)Pd(Ph)I 15 converted into 10 in 72% GC yield and 92% ee.
These results highlight the effect of the steric bulk of ImOx on its reactivity. First, the reductive elimination from a (ImOx)Pd(Ph)I complex is more favorable than the reverse process of oxidative addition. Second, the steric bulk prevents the direct insertion of the square-planar complex 15 into cyclohexenone, necessitating the abstraction of iodide by AgOTf to facilitate insertion by forming a cationic intermediate. A major difference of the catalytic conditions between ImOx and PyOx is the requirement for stoichiometric AgOTf to promote insertion, which proved to be the enantio-determining step.
We synthesized a series of ImOx-ligated palladium and nickel complexes to further characterize its structural and electronic properties in comparison to PyOx. Ligand substitution of (cod)PdCl_2_ (cod = 1,5-cyclooctadiene) with ImOx proceeded smoothly to deliver (^Dipp^Im^ iPr^Ox)PdCl_2_ 19 and (^Mes^Im^ tBu^Ox)PdCl_2_ 20 in good yields (SchemeA). Single-crystal X-ray diffraction reveals a square-planar geometry for 19, whereas the chloride adjacent to the tert-butyl group of ^Mes^Im^ tBu^Ox in 20 is slightly elevated, leading to a distortion from a square-planar geometry, possibly due to the steric hindrance. The bond lengths of C1–N1(imine) in (ImOx)PdCl_2_ 19 and 20 are significantly shorter than those of C1–N1(pyridine) in (^ tBu^PyOx)PdCl_2_. ?,? Additionally, the C1–C2 bonds, which connect the oxazoline and imine in 19 and 20, are substantially longer than those in (^ tBu^PyOx)PdCl_2_, indicating less extensive conjugation in (ImOx)PdCl_2_ compared to (^ tBu^PyOx)PdCl_2_. Although the bond lengths of Pd–N1 are comparable, the Pd–N2(oxazoline) bonds in (ImOx)PdCl_2_ 19 and 20 are shorter than those in (^ tBu^PyOx)PdCl_2_. Correspondingly, the Pd–Cl1 and Pd–Cl2 bond lengths in (ImOx)PdCl_2_ 19 and 20 are longer, suggesting a stronger trans-influence of ImOx relative to PyOx. However, the similar lengths of the Pd–Cl1 and Pd–Cl2 bonds indicate comparable trans-influences of the imine and oxazoline moieties.
Structural and Electronic Properties of ImOx Complexes
A preliminary investigation into the reactivity of ImOx in base-metal catalysis reveals its ability to promote the asymmetric reductive addition of aryl iodides to cyclohexenone with 92% ee (SchemeB), a reaction that produced nearly racemic products in previous studies.? The yield was low, likely due to the slow insertion of the nickel–aryl species into cyclohexenone, as evidenced by the formation of a significant amount of biphenyl byproduct. The addition of Lewis acids, such as AgOTf, improved the yield but decreased the ee. Notably, replacing ^Dipp^Im^ tBu^Ox with ^ tBu^PyOx led to no product formation.
While the yield using catalytic nickel requires further optimization, this preliminary result prompted us to evaluate the redox properties of ImOx, focusing on its ability to stabilize low-valent nickel radical complexes, which are critical for modern nickel-catalyzed cross-coupling reactions. Ligand substitution of Ni(cod)2 with ^Mes^Im^ iPr^Ox yielded a deep violet complex (^Mes^Im^ iPr^Ox)Ni(cod) 21. X-ray crystallography revealed an elongation of the C1–N1 and C2–N2 bonds and a shortening of C1–C2 bond compared to those in Pd(II) complexes, consistent with a reduction of the ImOx ligand. The ^1^H NMR spectrum of 21 reveals a resonance at 0.07 ppm, assigned to the methyl group adjacent to the imine, which is significantly shifted from its typical region around 2 ppm. DFT calculations of the electronic structure of 21 using the ORCA package resulted in a broken-symmetry BS(1,1) solution,? where a ligand-centered spin is antiferromagnetically coupled with a nickel-centered spin.? These data are consistent with the electronic structure of a Ni(I) coordinated with an ImOx radical anion. Chemical reduction of (^Dipp^Im^ tBu^Ox)Ni(Mes)Br 22 by KC_8_ resulted in an immediate color change from deep purple/red to light golden brown. The isotropic EPR signal of the reduced species 23 has a g value of 2.007, consistent with a ligand-centered radical but distinct from that of a free ligand radical anion (g = 2.000). DFT calculations of 23 further support the electronic structure of an ImOx-centered radical coordinated to a nickel(II).
When measuring the reduction potentials of ImOx by cyclic voltammetry (CV) under the same conditions as other common bidentate N-ligands, we found that ^Mes^Im^ tBu^Ox is more difficult to reduce than bpy, PyOx, and ADI, yet easier than bioxazoline (biOx) and pyridine-imidazole (PyIm) (SchemeC). However, when comparing the nickel-mesityl bromide complexes, (ImOx)Ni(Mes)Br displayed the least negative reduction potential. This data highlights ImOx’s pronounced tendency to exhibit redox activity in supporting nickel radical complexes, likely contributed by two factors. The imine moiety of ImOx lacks an α-O or N atom capable of providing resonance donation, as found in biOx or biIm, and its reduction does not require disrupting aromaticity, as is necessary with bpy or phen. The result that ImOx complexes facilitate reduction of coordinated metal species helps rationalize the difference in reactivity between ImOx and PyOx for the reductive 1,4-addition in SchemeB.
Conclusion
In summary, we developed a C 1-symmetric N,N-bidentate ligand, ImOx, derived from the chiral pool of amino acids. ImOx improved enantioselectivity in the palladium-catalyzed conjugate addition of phenylboronic acids to β-substituted α,β-unsaturated ketones. The steric effects at both the imine and oxazoline sites strongly influence the ee and be independently optimized owing to the modular structure of ImOx. Studies of well-defined organometallic intermediates demonstrate that the imine and oxazoline moieties in ImOx exhibit a comparable trans-influence, which is stronger than that of PyOx. Moreover, ImOx shows excellent redox activity, promoting the reduction of nickel complexes and stabilizing nickel radicals. We anticipate that ImOx will enrich the toolbox of chiral N-ligands for asymmetric catalysis.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Tamatam R.Shin D.Asymmetric Synthesis of US-FDA Approved Drugs over Five Years (2016 2020): A Recapitulation of Chirality Pharmaceuticals 202316333940010.3390/ph 1603033936986439 PMC 10052577 · doi ↗ · pubmed ↗
- 2Busacca C. A.Fandrick D. R.Song J. J.Senanayake C. H.The Growing Impact of Catalysis in the Pharmaceutical Industry Adv. Synth. Catal.201135311–121825186410.1002/adsc.201100488 · doi ↗
- 3Burk M. J.Gross M. F.Harper T. G. P.Kalberg C. S.Lee J. R.Martinez J. P.Asymmetric catalytic routes to chiral building blocks of medicinal interest Pure Appl. Chem.1996681374410.1351/pac 199668010037 · doi ↗
- 4Fernández-Pérez H.Etayo P.Panossian A.Vidal-Ferran A.Phosphine–Phosphinite and Phosphine–Phosphite Ligands: Preparation and Applications in Asymmetric Catalysis Chem. Rev.201111132119217610.1021/cr 100244 e 21250669 · doi ↗ · pubmed ↗
- 5Pfaltz A.Chiral semicorrins and related nitrogen heterocycles as ligands in asymmetric catalysis Acc. Chem. Res.199326633934510.1021/ar 00030 a 007 · doi ↗
- 6Tasker S. Z.Standley E. A.Jamison T. F.Recent advances in homogeneous nickel catalysis Nature 201450929930910.1038/nature 1327424828188 PMC 4344729 · doi ↗ · pubmed ↗
- 7Cherney A. H.Kadunce N. T.Reisman S. E.Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents To Construct C–C Bonds Chem. Rev.20151159587965210.1021/acs.chemrev.5b 0016226268813 PMC 4566132 · doi ↗ · pubmed ↗
- 8Choi J.Fu G. C.Transition metal–catalyzed alkyl-alkyl bond formation: Another dimension in cross-coupling chemistry Science 20173566334 eaaf 723010.1126/science.aaf 723028408546 PMC 5611817 · doi ↗ · pubmed ↗