Case Report: Type II Bartter syndrome with a novel KCNJ1 variant in a premature neonate presenting with features of salt-wasting congenital adrenal crisis and pseudo-hypoaldosteronism
Heung-Ching Tsui, Hua Tse-Timothy Cheng, Kai-Yee Lam, Lai-Ting Leung, Ka-Nam Au, Wai-Yu Wong, Luen Yee-Sylvia Siu, Lap-Ming Wong

TL;DR
A premature baby with a rare kidney disorder was misdiagnosed initially but later found to have a new genetic variant causing Bartter syndrome type II.
Contribution
A novel KCNJ1 gene variant was identified in a neonate with Bartter syndrome type II, aiding in early diagnosis and management.
Findings
A premature neonate presented with adrenal crisis-like symptoms but was later diagnosed with Bartter syndrome type II.
Compound heterozygous missense variants in the KCNJ1 gene were identified through whole exome sequencing.
Treatment with indomethacin showed favorable outcomes after initial fludrocortisone therapy.
Abstract
Bartter syndrome (BS) is a rare group of inherited renal tubulopathies. Diagnosis of BS type II is challenging in the neonatal period as its clinical findings and biochemical features may mimic that of adrenal crisis and pseudo-hypoaldosteronism (PHA) initially. Treatment should be instituted immediately for acute adrenal insufficiency as it is a medical emergency, then modified according to available investigation results and treatment response. We describe a premature female neonate with an antenatal history of severe unexplained polyhydramnios, presented with features of adrenal crisis managed with hydrocortisone and fludrocortisone. Initial endocrine investigations excluded salt-wasting congenital adrenal hyperplasia (SW-CAH) and pointed to the diagnosis of PHA with hyperreninemic hyperaldosteronism. Hydrocortisone was gradually weaned off while fludrocortisone was continued for…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
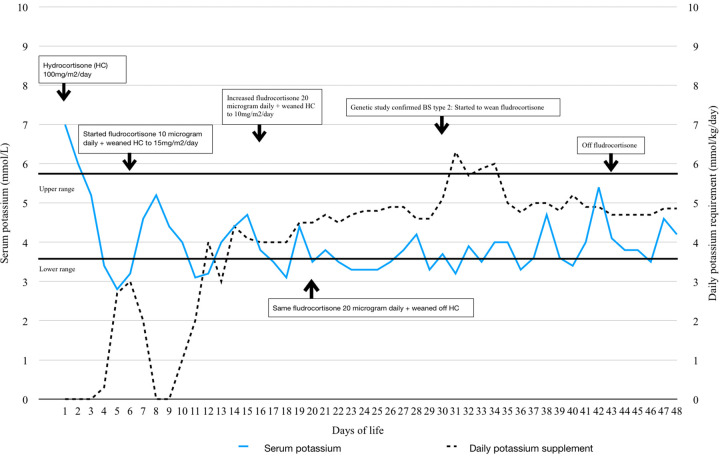 Figure 1
Figure 1| Investigation | Results | Normal range |
|---|---|---|
| Spot cortisol | 481 nmol/L | 101–536 nmol/L |
| ACTH | 1.4 pmol/L | ≤10.2 pmol/L |
| 17-hydroxyprogesterone | 3 nmol/L | <8 nmol/L |
| Androstenedione | 1.1 nmol/L | 0.7–10.1 nmol/L |
| Testosterone | <0.2 nmol/L | <0.8 nmol/L |
| Renin | 284.88 ng/ml/h | 2–35 ng/ml/h |
| Aldosterone | 11,440 pmol/L | ≤6,025 pmol/L |
| Normal or low blood pressure | Differential diagnosis |
|---|---|
| Metabolic alkalosis | |
| Low urine chloride |
Congenital chloride diarrhoea Chloride deficient diet Hypercapnea Pseudo-bartter syndrome e.g., in cystic fibrosis |
| High urine chloride |
Bartter syndrome (with hypercalciuria) Gitelman syndrome (with hypocalciuria) Diuretic use |
| Metabolic acidosis |
Renal tubular acidosis Fanconi syndrome |
| High blood pressure | Differential diagnosis |
| High renin |
Renal artery stenosis Renal parenchymal disease Renal tumour/pheochromcytoma |
| Low renin and high aldosterone |
Primary hyperaldosteronism |
| Low renin and low aldosterone |
Exogenous mineralocorticoid/glucocorticoid 17 alpha hydroxylase deficiency 17 beta hydroxylase deficiency Liddle syndrome |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsIon Transport and Channel Regulation · Electrolyte and hormonal disorders · Ion channel regulation and function
Introduction
1
Bartter syndrome (BS) is a group of inherited salt-losing renal tubulopathies characterized by impaired salt reabsorption in the thick ascending limb (TAL) of the loop of Henle with activation of the renin-angiotensin system, resulting in polyuria, hypokalemia, hypochloremic metabolic alkalosis and normotensive hyperreninemic hyperaldosteronism (1).
Acute adrenal insufficiency is rare in the neonatal period and salt-wasting congenital adrenal hyperplasia (SW-CAH) is the commonest cause of it (2). Adrenal crisis is suspected when newborns present with hyperkalemia with hypotension. Life-saving treatment of adrenal crisis should be initiated immediately. In contrast to other types of BS, BS type II may present with transient hyperkalemia in the neonatal period, mimicking SW-CAH and pseudo-hypoaldosteronism (PHA) type I until urine tests and serum renin and aldosterone are available. Genetic study should be done early to get a definitive diagnosis and guide the subsequent management. Before the genetic result is available, treatments should be modified according to the contemporaneous biochemical results and clinical progress.
In this case report, we describe a premature female neonate presented with clinical features of acute adrenal insufficiency. The clinical condition and biochemical parameters were stabilized after the initiation of treatment for adrenal crisis. The patient developed polyuria and hypokalemia at 1 week of age. Initial endocrine investigations excluded SW-CAH and pointed to the diagnosis of PHA type I with markedly elevated serum renin and aldosterone. However, the urine potassium was suggestive of salt losing which should not be present in PHA type I. Genetic test confirmed the diagnosis of BS type II with compound heterozygous likely pathogenic variants identified in the KCNJ1 gene. The management approach was then refined according to the genetic results.
Case presentation
2
General information
2.1
A baby girl was delivered at gestation 29 + 4 weeks by crash Caesarean section due to fetal distress with a birth weight of 1.27 kg which was appropriate to gestational age. Apgar score was 6 at 1 min and 7 at 5 min of life.
This was the first pregnancy of a non-consanguineous Chinese couple. Mother had seronegative rheumatoid arthritis with stable control on sulphasalazine. There was no family history of endocrine or renal diseases. Antenatal ultrasonography revealed significant polyhydramnios since 24 weeks of gestation. Amniotic Fluid Index (AFI) was 47.5 cm with a normal stomach bubble. There was no gestational diabetes mellitus and the morphology scan was unremarkable. Amnioreduction was performed at 25 weeks of gestation with antenatal steroid given. Chromosome microarray on the amniotic fluid was normal.
After delivery, the baby required transient invasive ventilatory support. Neonatal sepsis workup was negative. There was hypotension requiring saline boluses and dopamine infusion for less than 1 day. Newborn examination showed no dysmorphism and she had normal female genitalia with no signs of virilization.
Diagnostic assessment
2.2
At 20 h of life, routine blood test showed severe hyperkalemia with serum potassium level of 7 mmol/L (reference range 3.7–5.9 mmol/L) and hyponatremia with serum sodium level of 131 mmol/L (reference range 133–146 mmol/L) (Figure 1). Serum urea was raised at 10.9 mmol/L (reference range 1–8.2 mmol/L). Serum creatinine was normal. Plasma glucose was 2.9 mmol/L. There was no significant metabolic acidosis nor alkalosis with gas showing pH 7.4, bicarbonate 19.3 mmol/L (reference range 22–26 mmol/L) and base excess −4 mmol/L (reference range −2 to −3 mmol/L). There was mild tachycardia with heart rate 180/min. Blood pressure was normal.
Trend of serum potassium, daily potassium supplement, hydrocortisone and fludrocortisone dosage.
The presumptive diagnosis was acute adrenal insufficiency. Endocrine investigations including cortisol, adrenocorticotropic hormone (ACTH), 17-hydroxyprogesterone (17-OHP) and androgen profile were performed. Then, she was treated immediately with fluid resuscitation and intravenous hydrocortisone at 100 mg/m^2^/day and was put on fludrocortisone later. Dextrose-insulin drip was given for 1 day and her hyperkalemia promptly settled (Figure 1). She also had polyuria with urine output up to 8 ml/kg/h since day 1 of life with significant weight loss of 20%. Despite hyperkalemia, the urine potassium was 5 mmol/L and the fractional excretion of potassium (FeK) was 10% which spoke against hypoaldosteronism.
The serum potassium dropped to 2.8 mmol/L since day 5 of life. There was hypochloremic metabolic alkalosis with serum chloride 96 mmol/L (reference range 98–113 mmol/L). The urine biochemistry was suggestive of salt wasting and concentration disability with spot urine potassium of 8.7 mmol/L, FeK 19.9%; Fractional excretion of sodium (FeNa) 9.5%; urine chloride of 81 mmol/L, fractional excretion of chloride 11% and urine osmolality of 257 mOsm/kg. She also had hypercalciuria with urine calcium to creatinine ratio of 5.34 mmol/mmol. Meanwhile, endocrine investigations were all unremarkable except markedly elevated renin and aldosterone (Table 1). Newborn metabolic screening also excluded SW-CAH.
Genetic testing
2.3
Buccal swab together with parental blood samples were then sent for urgent trio Whole Exome Sequencing (WES) on day 12 of life for genes related to renal tubulopathies, fetal anomalies and aldosterone disorders. It was performed by Roche KAPA HyperExome using Illumina NextSeq550. Target mean sequencing depth was 100× and a minimum of 99% of target bases were sequenced to at least 20×. Single nucleotide variants, small insertion-deletion variants (≦15 bp) and copy number variants, which were located in exons and 10 bp of adjacent intronic sequences were targeted for curation. Exome analysis detected compound heterozygous missense variants in exon 3 of KCNJ1 (NM_153766.3) gene: the maternally-inherited c.598A>C p.(Ser200Arg) variant and the novel paternally-inherited c.589C>T p. (Leu197Phe) variant. Both variants were classified as likely pathogenic and diagnosis of BS type II was confirmed on day 30 of life.
Treatment and outcome
2.4
Indomethacin was started at corrected 37 gestation weeks, stepping up the dose from 1 mg/kg/day to 3 mg/kg/day in 2 weeks. Sodium and potassium supplements were tapered off at 3 months old with normal electrolytes and renal function. Hypercalciuria improved with the latest urine calcium to creatinine ratio at 0.85 mmol/mmol. Ultrasonography of the kidneys showed medullary nephrocalcinosis. Fractional excretion of sodium and potassium reduced to 0.9% and 9.5% respectively. Serum creatinine remained normal. Serum renin and aldosterone levels also normalized. Upon follow up, she had satisfactory weight gain.
Discussion
3
Pathophysiology of BS type II
3.1
BS has an estimated incidence of 0.1 in 100,000 individuals. To date, five genes have been identified for various phenotypic presentations of BS. BS type II accounts for 19% of BS according to a systematic review (3). BS type II is an autosomal recessive disorder by the mutation of the KCNJ1 gene. This gene encodes the apical renal outer medullary potassium channel (ROMK) which allows recycling of potassium into the tubular lumen in the TAL of the loop of Henle. The decreased recycling of potassium impairs the function of the sodium-potassium-chloride cotransporter which is responsible for sodium, potassium and chloride reabsorption, resulting in salt losing and decreased medullary interstitial hypertonicity. Thus, the water reabsorption in the collecting duct is impaired resulting in isothenuria. The impaired reabsorption of sodium chloride and potassium leads to decrease of tubular lumen positivity which is important in calcium and magnesium reabsorption. Therefore, calcium and magnesium loss in urine are features of BS type II. Hypercalciuria can lead to nephrocalcinosis which is common in BS type II (4). ROMK is also present in the macula densa in the juxta-glomerulus apparatus which, in normal function, detects the sodium chloride concentration in the distal convoluted tubule. Despite sodium chloride loss in the urine, the faulty ROMK in the macula densa fails to sense it, resulting in the release of prostaglandin and paradoxical activation of the renin-angiotensin system (RAS).
BS type II is classically presented with hyperkalemia then hypokalemia which mimics PHA type I on first presentation. One proposed mechanism of transient postnatal hyperkalemia in BS type II is the decreased Na-K-ATPase activity related to prematurity, causing a shift of potassium to the extracellular space. ROMK is also expressed in the cortical collecting duct, responsible for potassium secretion. Inactivation of its function reduces urinary potassium secretion and results in transient hyperkalemia (5). With the gradual maturation of the Na-K-ATPase activity, hyperkalemia resolves and evolves into hypokalemia. The role of potassium secretion of ROMK in the cortical collecting duct also explains the more easily corrected hypokalemia in BS type II.
Clinical presentations and differential diagnoses of BS type II
3.2
Antenatally, BS presents commonly with severe polyhydramnios and prematurity. Any unexplained severe polyhydramnios should raise suspicion of BS. Genetic tests should be offered or amniotic fluid for Bartter index may be used if genetic tests are not available (6).
Early recognition of BS type II remains challenging in the neonatal period. Our case initially presented like adrenal crisis. The most common cause is SW-CAH which requires targeted endocrine investigations and prompt treatments. With the availability of test results and treatment response, the list of differential diagnoses can then be narrowed down. SW-CAH in a non-virilized female neonate can be due to lipoid CAH, P450 cytochrome side chain cleavage deficiency and 3β-hydroxysteroid dehydrogenase deficiency (7). These were excluded after cortisol and ACTH came back normal. PHA type I then became the likely differential diagnosis of hyperreninemic hyperaldosteronism. However, the development of hypokalemia in our patient with high FeK and metabolic alkalosis spoke against it.
Importantly, hyperkalemia quickly transits into hypokalemia in BS type II. There are few case reports describing hyperkalemia, instead of hypokalemia in neonates with BS type II. Finer et al. reported in a case series involving 12 neonates with genetically confirmed BS type II having significant salt-wasting. The mean of the peak potassium level was 9 mmol/L and the mean of the trough sodium level at 124 mmol/L. Hyperkalemia in this group appeared on day 3 of life and normalized by the end of the first week (8). Nozu et al. also illustrated a case with hyperkalemia, hyponatremia and hyperreninemic hyperaldosteronism in the early neonatal period. This patient was also initially diagnosed as PHA until genetic study confirmed homozygous mutation of the KCNJ1 gene (9). When hypokalemia occurs, focused investigations should be performed to evaluate its cause. In our case, there was evidence of urinary loss of potassium, normal blood pressure, metabolic alkalosis and markedly elevated serum renin and aldosterone. Differential diagnoses include congenital chloride diarrhea and pseudo-bartter syndrome which were excluded by a fractional excretion of chloride higher than 0.5% (10, 11). The presence of hypercalciuria and normal magnesium level distinguish BS from Gitelman syndrome.
We illustrate that biochemical findings can overlap between SW-CAH, PHA and BS type II in the early neonatal period due to transient hyperkalemia. Clinicians can use blood pressure, gas, serum and paired urine electrolytes to establish the diagnosis in cases of potassium disturbance (Table 2).
Genetic diagnosis of BS
3.3
The European Rare Kidney Disease Reference Network Working Group (ERKNet Working Group) for Tubular Disorders recommends genetic testing for all patients with clinical suspicion of BS. BS type I to IV is autosomal recessively inherited and BS type V is X-linked recessively inherited. The detection of inactivating mutations in both alleles confirms the diagnosis of recessive forms of BS. The sensitivity and specificity of the current genetic test is 75% and 100% respectively (12).
For our case, a likely pathogenic variant of KCNJ1 gene [c.598A>C p. (Ser200Arg)] was detected from the maternally inherited allele. This variant is absent in control population in gnomAD. However, a different variant (c.600C>G and c.600C>A)) giving rise to the same amino acid change has been reported to be pathogenic in patients with clinical features of BS (PMID: 8841184. 24400161. 29942493. ClinVar VCV001995667.2. VCV000009155.2). Experimental evidence and functional studies also suggest that p.(Ser200Arg) change affects potassium channel activity and function (PMID: 8841184. 9580661. 2440161). REVEL score is 0.926. Therefore, this variant is considered to be likely pathogenic. The KCNJ1 gene [c.589C>T p. (Leu197Phe)] was detected from the paternally inherited allele. This variant has not been reported in human mutation databases and literature so far. REVEL score is 0.774 fulfilling a moderate score for in-silico prediction according to American College of Medical Genetics (ACMG) criteria. Both variants are absent in control populations in gnomAD. These two variants are only 3 amino acids apart and are situated in the potassium channel inwardly rectifying (Kir) cytoplasmic COOH-termini protein domain (PMID: 35463019). Our case demonstrated classical features of BS type II with specific clinical and biochemical phenotypes, caused by compound heterozygous KCNJ1 variants c.598A>C p. (Ser200Arg) and c.589C>T p. (Leu197Phe). Whole exome sequencing did not detect any other clinically significant variants, reducing the possibility of other differential diagnoses that were initially suspected.
Treatments
3.4
Initial treatment includes supplementation with sodium chloride and potassium chloride. Electrolyte supplements should be spread throughout the day to minimize the fluctuation of plasma level. Excessive salts will exaggerate the polyuria and lead to detrimental secondary bladder dysfunction or refluxing diseases. Therefore, judicious use of salt supplements with realistic targets can be made for patients with BS. Patients with BS type II may develop secondary nephrogenic diabetes insipidus and chronic use of sodium supplementation can aggravate hypernatremic dehydration (13). Non-Steroidal Anti-Inflammatory Drug (NSAID) is recommended for all BS to suppress cyclooxygenase and prostaglandin production, leading to decreased activation of the RAS. The ERKNet Working Group suggests that patients may develop “tolerance” to NSAID over time (12) so its use should be individualized to biochemical and clinical response. During follow up, it is crucial to monitor the growth, hydration, serum and urine biochemistry and development of nephrocalcinosis.
Our patient was started on indomethacin reaching full dosage of 3 mg/kg/day. The RAS was suppressed with improvement of serum and urinary excretion of electrolytes.
Conclusion
4
Clinical and biochemical features of SW-CAH and PHA type I mimic that of BS type II with salt wasting in the early neonatal period, making the diagnostic process challenging. Acute adrenal insufficiency is rare in the neonatal period. Treatment should be instituted before investigation results are available. The presence of unexplained antenatal polyhydramnios and polyuria prompts targeted investigations of renal causes of salt-losing picture. Genetic tests provide a definitive diagnosis of BS and can guide subsequent management and genetic counselling. The typical clinical features of our patient suggest the KCNJ1[c. 589C>T p. (Leu197Phe)] is a novel likely pathogenic variant.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Florea L Caba L Gorduza EV. Genetic heterogeneity in Bartter syndrome: clinical and practical importance. Front Pediatr. (2022) 10:908655. 10.3389/fped.2022.90865535722471 PMC 9203713 · doi ↗ · pubmed ↗
- 2Buonocore F Mc Glacken-Byrne SM Del Valle I Achermann JC. Current insights into adrenal insufficiency in the newborn and young infant. Front Pediatr. (2020) 8:619041. 10.3389/fped.2020.61904133381483 PMC 7767829 · doi ↗ · pubmed ↗
- 3Qasba RK Bucharles ACF Piccoli MVF Sharma P Banga A Kamaraj B Bartter syndrome: a systematic review of case reports and case series. Medicina (Kaunas). (2023) 59(9):1638. 10.3390/medicina 5909163837763757 PMC 10537044 · doi ↗ · pubmed ↗
- 4Welling PA Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol. (2009) 297(4):F 849–63. 10.1152/ajprenal.00181.200919458126 PMC 2775575 · doi ↗ · pubmed ↗
- 5Sato K Kondo T Iwao H Honda S Ueda K. Internal potassium shift in premature infants: cause of nonoliguric hyperkalemia. J Pediatr. (1995) 126(1):109–13. 10.1016/s 0022-3476(95)70511-27815197 · doi ↗ · pubmed ↗
- 6Vedmedovska N Luse L Rezeberga D Miskova A Rasnaca K. VP 32.08: clinical case of pregnancy and follow-up of bartter syndrome (type 2) with a novel mutation. Ultrasound Obstet Gynecol. (2020) 56(S 1):190. 10.1002/uog.22811 · doi ↗
- 7Balsamo A Baronio F Ortolano R Menabo S Baldazzi L Di Natale V Congenital adrenal hyperplasias presenting in the newborn and young infant. Front Pediatr. (2020) 8:593315. 10.3389/fped.2020.59331533415088 PMC 7783414 · doi ↗ · pubmed ↗
- 8Finer G Shalev H Birk OS Galron D Jeck N Sinai-Treiman L Transient neonatal hyperkalemia in the antenatal (ROMK defective) Bartter syndrome. J Pediatr. (2003) 142(3):318–23. 10.1067/mpd.2003.10012640382 · doi ↗ · pubmed ↗