Systemic Lupus Erythematosus-Associated Thrombotic Thrombocytopenic Purpura: A Case Report
Ounci Es-Saad, Adil Zyani, Ayman Bouchlaghem, Rajae Alkouh, Smael Labib

TL;DR
A rare case of thrombotic thrombocytopenic purpura in a lupus patient was successfully treated with plasmapheresis and immunosuppressive therapy.
Contribution
This case report adds to the limited literature on SLE-associated TTP and emphasizes the importance of early diagnosis and treatment.
Findings
ADAMTS13 deficiency confirmed the diagnosis of TTP in a patient with SLE.
Plasmapheresis and immunosuppressive therapy led to significant clinical and laboratory improvement.
Early recognition of TTP in SLE patients with neurological symptoms is crucial for favorable outcomes.
Abstract
Systemic lupus erythematosus-associated thrombotic thrombocytopenic purpura (SLE-TTP) is a rare but life-threatening condition that requires prompt recognition and treatment. We report a case of a patient with systemic lupus erythematosus (SLE) who presented with encephalopathy and was subsequently diagnosed with thrombotic thrombocytopenic purpura (TTP) based on ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin motifs 13) deficiency. The patient was successfully treated with plasmapheresis. A 42-year-old woman with a history of SLE presented with febrile encephalopathy and was admitted to the intensive care unit (ICU). Laboratory evaluation revealed microangiopathic hemolytic anemia and severe thrombocytopenia. MRI showed leptomeningeal enhancement and white matter changes suggestive of neuro-lupus. However, ADAMTS13 activity was <1% with detectable anti-ADAMTS13…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
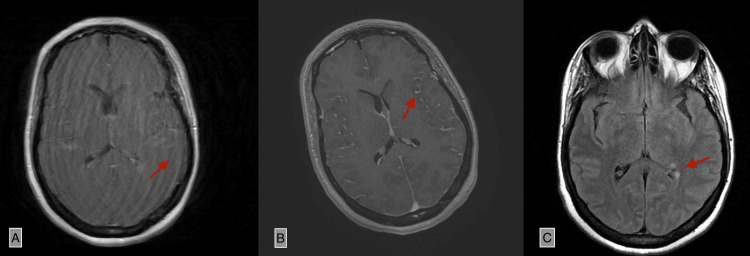 Figure 1
Figure 1| Parameter | Value | Reference Range |
| Hemoglobin | 6.4 g/dL | 11-14 g/dL |
| Hematocrit | 18.9% | 36-48% |
| MCV | 108.8 fL | 80-100 fL |
| MCH | 37.1 Pg | 27-33 pg |
| MCHC | 34.1 g/dL | 32-36 g/dL |
| Platelets | 8000/uL | 150000-400000/uL |
| Reticulocytes | 446100/mm³ | 25000-75000/mm³ |
| Schistocytes | 2.6% | <1% |
| LDH | 993 U/L | 140-280 U/L |
| Haptoglobin | <0.1 g/L | 0.3-2.0 g/L |
| Total WBC Count | 17270/uL | 4000-11 000/uL |
| Neutrophils | 13310/uL | 2000-7500/uL |
| Lymphocytes | 3120/uL | 1000-4800/uL |
| CRP | 22 mg/L | <5 mg/L |
| AST | 122 U/L | 10-40 U/L |
| ALT | 89 U/L | 7-56 U/L |
| GGT | 100 U/L | 9-48 U/L |
| Alkaline Phosphatase | 94 U/L | 30-120 U/L |
| Total Bilirubin | 3.8 mg/L | 0.3-1.2 mg/dL |
| Direct Bilirubin | 1 mg/L | 0-0.3 mg/dL |
| Indirect Bilirubin | 2.8 mg/L | 0.1-1.0 mg/dL |
| Hepatitis A Serology | Negative | Negative |
| HIV | Negative | Negative |
| Sodium | 141 mmol/L | 135-145 mmol/L |
| Potassium | 4.2 mmol/L | 3.5-5.0 mmol/L |
| Chloride | 100 mmol/L | 98-107 mmol/L |
| Calcium | 87 mg/L | 85-105 mg/L |
| Vitamin B12 | 230 pg/mL | 200-900 pg/mL |
| Folate (B9) | 2.9 ng/mL | 3-20 ng/mL |
| Urea | 0.50 g/L | 0.17-0.43 g/L |
| Serum Creatinine | 7.7 mg/L | 0.6-1.2 mg/dL |
| Coombs Test | Negative | Negative |
| Parameter | Value | Reference Range |
| ADAMTS13 Activity | <1% | >67% |
| Anti-ADAMTS13 Antibodies | >15 U/mL Positive | Negative |
| Anti-cardiolipin IgG/IgM | Negative | Negative |
| Anti-Beta2 Glycoprotein 1 IgG/IgM | Negative | Negative |
| Anti-DNA | Negative | Negative |
| Lupus Anticoagulant | Negative | Negative |
| Complement C3 | 1.07 g/L | 0.9-1.8 g/L |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsComplement system in diseases · Renal Diseases and Glomerulopathies · Platelet Disorders and Treatments
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease with multi-organ involvement, ranging from mild mucocutaneous manifestations to life-threatening organ dysfunction. Hematologic manifestations of SLE include autoimmune hemolytic anemia, leukopenia, lymphopenia, and thrombocytopenia. Thrombotic thrombocytopenic purpura (TTP), while not a classical hematologic feature of SLE, may occur as a rare and severe complication. TTP is a subtype of thrombotic microangiopathy (TMA) that is exceedingly rare but potentially fatal [1,2].
TTP is characterized by thrombocytopenia, microangiopathic hemolytic anemia, and organ dysfunction. It is an autoimmune disorder mediated by antibodies against ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin motifs 13), a von Willebrand factor-cleaving protease, leading to its severe functional deficiency [3]. Diagnosis is often based on the presence of microangiopathic hemolytic anemia and thrombocytopenia, with or without renal, neurologic, or fever-related symptoms. In clinical practice, tools such as the PLASMIC score are sometimes used to assess the likelihood of TTP, especially in settings where access to ADAMTS13 testing is delayed.
The overlap between SLE and TTP, referred to as systemic lupus erythematosus-associated thrombotic thrombocytopenic purpura (SLE-TTP), poses a significant diagnostic challenge due to the shared clinical features, including fever, renal involvement, and neuropsychiatric symptoms [4,5]. While primary (idiopathic) TTP is typically isolated, SLE-TTP occurs in the context of systemic autoimmunity, often complicating disease flares and presenting with overlapping laboratory and imaging findings. Although classically TTP was diagnosed clinically based on a pentad of findings, modern approaches rely heavily on laboratory confirmation, with an ADAMTS13 activity level <10% being diagnostic. Prompt initiation of plasma exchange and immunosuppressive therapy is essential for favorable outcomes [3,6].
This case highlights the diagnostic ambiguity between neuropsychiatric lupus and TTP in the setting of acute encephalopathy. It also underscores the importance of early ADAMTS13 testing and rapid initiation of combined immunosuppressive and plasma exchange therapy, which contributed to a favorable neurologic recovery.
Case presentation
A 42-year-old woman with a known history of cutaneous and articular SLE, treated with hydroxychloroquine for one year, was admitted with febrile encephalopathy. She presented with jaundice, asthenia, and progressive altered mental status. On examination, she was febrile (38°C), hemodynamically stable, and had a fluctuating Glasgow Coma Score between nine and 12. Generalized jaundice and hepatosplenomegaly were noted.
Initial laboratory investigations were concerning for microangiopathic hemolytic anemia, evidenced by anemia, reticulocytosis, the presence of schistocytes on peripheral smear, elevated lactate dehydrogenase (LDH), and undetectable haptoglobin. The patient also had profound thrombocytopenia and elevated inflammatory markers, while renal function and electrolyte levels were within normal limits (Table 1).
Cardiac evaluation by transthoracic echocardiography showed no abnormalities. Brain MRI revealed diffuse leptomeningeal enhancement and periventricular white matter hyperintensities with contrast enhancement, initially suggestive of neuropsychiatric lupus or CNS vasculitis (Figure 1).
Contrast-enhanced MRI showing diffuse supratentorial leptomeningeal thickening and hyperintense signals in the left periventricular white matter, with post-contrast enhancement. These findings are suggestive of lupus-related vascular involvement with leptomeningitis(A) Axial T1-weighted post-contrast sequence showing periventricular contrast enhancement (arrow). (B) Axial T1-weighted post-contrast sequence demonstrating diffuse supratentorial leptomeningeal enhancement (arrow). (C) Axial FLAIR sequence showing periventricular hyperintense signal (arrow).
The PLASMIC score was calculated at six, placing the patient in a high-risk category for severe ADAMTS13 deficiency. ADAMTS13 activity was subsequently found to be <1%, with positive anti-ADAMTS13 antibodies, confirming the diagnosis of acquired TTP. Autoimmune workup showed negative antiphospholipid antibodies (anti-cardiolipin and anti-β2 glycoprotein I), anti-dsDNA, and lupus anticoagulant. Complement C3 was within the normal range (Table 2).
The patient was treated with four sessions of plasmapheresis, high-dose corticosteroids (methylprednisolone 1 g daily for three days followed by oral prednisone at 1 mg/kg/day), a single dose of rituximab (900 mg), one dose of cyclophosphamide (0.5 g/m²), and supportive measures including anti-epileptic therapy. Improvement in mental status and hematologic parameters was noted after the first session of plasmapheresis. She gradually regained full consciousness (GCS 15) and was transferred to the internal medicine department after 17 days, although segmental muscle weakness persisted - graded at 4/5 in the upper limbs and 2/5 in the lower limbs.
She remained hospitalized in the internal medicine unit for 15 additional days, during which she continued to show clinical improvement. Muscle strength gradually recovered, and biological parameters further improved. At discharge, laboratory findings showed significant recovery, with hemoglobin rising to 10.8 g/dL, platelet count to 278,000/uL, LDH down to 331 U/L, and CRP reduced to 1 mg/L. Renal function remained stable.
Discussion
SLE is a complex autoimmune disease with a wide spectrum of clinical manifestations. Among its many complications, TTP remains rare but clinically significant due to its potential severity. TTP is a TMA characterized by severe ADAMTS13 deficiency leading to the accumulation of ultra-large von Willebrand factor (ULVWF) multimers and subsequent microvascular platelet-rich thrombi formation [1]. This results in ischemic injury, particularly affecting the CNS and kidneys.
The incidence of TTP is estimated at 3.7 per million annually, with a higher frequency in young women and individuals with autoimmune backgrounds [2]. In patients with SLE, the prevalence of TTP ranges from 1% to 4% [3,4]. The SLE-TTP presents unique diagnostic and therapeutic challenges due to overlapping features such as fever, thrombocytopenia, hemolytic anemia, renal dysfunction, and neuropsychiatric symptoms [5].
In our case, the diagnosis of immune-mediated TTP (iTTP) was confirmed by the presence of schistocytes, an ADAMTS13 activity <1%, and anti-ADAMTS13 autoantibodies. While ADAMTS13 testing is confirmatory, the PLASMIC score remains a valuable clinical tool for early recognition. This score evaluates parameters including platelet count, hemolysis markers, INR, creatinine, and the presence of active cancer or transplant history. Our patient’s score was six, categorizing her as high risk with a 72% chance of severe ADAMTS13 deficiency, thus justifying urgent plasma exchange even before confirmatory assays were available [6].
ADAMTS13 deficiency causes the accumulation of ULVWF, which promotes spontaneous platelet aggregation and the formation of microthrombi in small vessels. This leads to end-organ ischemia, particularly in the CNS, where patients often present with altered mental status, seizures, or stroke-like symptoms [1,7]. The MRI findings in our case, including leptomeningeal enhancement and periventricular white matter changes, could represent either neuropsychiatric lupus (NPSLE) or TTP-related microvascular ischemia. Distinguishing the two is critical but challenging. Petz et al. previously highlighted that CNS involvement is often prominent in both disorders and can mimic each other [8]. However, our patient's rapid neurological improvement following plasma exchange and immunosuppression supports TTP as the primary pathology.
Febrile encephalopathy with thrombocytopenia has a broad differential diagnosis. Tick-borne illnesses such as anaplasmosis, ehrlichiosis, and babesiosis can present similarly and should be considered, especially in endemic regions [9]. In particular, Anaplasma phagocytophilum has been reported to cause encephalitis and TMA-like syndromes. Blood smear review and PCR assays are essential to exclude these infections prior to initiating immunosuppression.
Patients with SLE-TTP often demonstrate a phenotype of severe thrombocytopenia, prominent CNS involvement, and relatively mild renal dysfunction, which was consistent with our case. Yue et al. found that patients with SLE-TTP and ADAMTS13 inhibitor positivity had significantly better outcomes and lower mortality than those with primary TTP [1]. This paradoxically favorable prognosis in SLE-TTP may be related to earlier diagnosis and more aggressive immunosuppressive treatment [1,4].
Our patient was promptly treated with plasma exchange and high-dose corticosteroids, resulting in rapid hematologic and neurological recovery. The cornerstone of TTP therapy remains daily therapeutic plasma exchange (TPE), which removes the inhibitor and replenishes ADAMTS13. Adjunctive immunosuppression is essential to halt autoantibody production. In refractory or relapsing TTP, rituximab has shown efficacy by depleting B-cells, with some authors advocating for its use in the first episode of iTTP when ADAMTS13 inhibitors are detected [10,11]. In our case, rituximab was initiated early due to the autoimmune nature of the TTP and concern for severe inhibitor-mediated disease.
Cyclophosphamide was also administered, given the coexisting active lupus and potential overlap with NPSLE. Although controversial, some authors suggest that cyclophosphamide may aid in treating SLE-associated TMA by targeting autoreactive lymphocytes [12]. As the patient was a young woman, fertility preservation was discussed prior to administration, and gonadoprotective measures were considered.
Eculizumab, a terminal complement inhibitor, has been reported in multiple case series to be effective in patients with SLE-associated TMA refractory to plasma exchange and steroids. Its role in cases with features of complement activation or overlap with hemolytic uremic syndrome (aHUS) is under investigation [13,14]. While our patient responded to conventional therapy, complement blockade may be considered in future relapses or refractory scenarios.
The prognosis of SLE-TTP has historically been poor, with mortality rates exceeding 50% prior to the advent of plasma exchange [3]. More recent series, however, report survival rates above 80% with prompt diagnosis and aggressive therapy [1,4,11]. Long-term follow-up is essential due to relapse risk, which can occur in up to 40% of patients [15].
This case contributes to the limited but growing literature on CNS-predominant SLE-TTP. The patient's rapid neurological recovery and hematological response underscore the importance of early recognition and timely initiation of plasma exchange and immunosuppressive therapy.
Conclusions
The SLE-TTP is rare, yet it must be considered in lupus patients presenting with unexplained neurological symptoms, cytopenias, and laboratory evidence of hemolysis. Clinical tools such as the PLASMIC score can aid in early identification, particularly in emergency settings where ADAMTS13 testing is delayed. Nonetheless, ADAMTS13 activity measurement remains essential for definitive diagnosis and therapeutic guidance. This case demonstrates that early recognition and prompt initiation of plasma exchange, alongside corticosteroids and immunosuppressive therapy, can lead to rapid neurological improvement, resolution of hemolysis, and normalization of platelet counts. Future multicenter studies are needed to better define the clinical spectrum, treatment strategies, and long-term outcomes in patients with SLE-TTP.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Immune-mediated thrombotic thrombocytopenic purpura in patients with and without systemic lupus erythematosus: a retrospective study Orphanet J Rare Dis Yue C Su J Fan X 2251520203285923710.1186/s 13023-020-01510-9PMC 7456051 · doi ↗ · pubmed ↗
- 2Acquired idiopathic ADAMTS 13 activity deficient thrombotic thrombocytopenic purpura in a population from Japan P Lo S One Matsumoto M Bennett CL Isonishi A 07201210.1371/journal.pone.0033029 PMC 329972722427934 · doi ↗ · pubmed ↗
- 3Review of thrombotic thrombocytopenic purpura in the setting of systemic lupus erythematosus Semin Arthritis Rheum Musio F Bohen EM Yuan CM 119281998972633110.1016/s 0049-0172(98)80023-1 · doi ↗ · pubmed ↗
- 4A systematic review of the role of eculizumab in systemic lupus erythematosus-associated thrombotic microangiopathy BMC Nephrol Wright RD Bannerman F Beresford MW Oni L 245212020 https://doi.org/10.1186/s 12882-020-01888-53260554010.1186/s 12882-020-01888-5PMC 7329551 · doi ↗ · pubmed ↗
- 5Clinical features and prognosis of patients with thrombotic thrombocytopenic purpura associated with systemic lupus erythematosus: a review of 25 cases Ital J Pediatr Li J Jiang JJ Wang CY 55452019 https://doi.org/10.1186/s 13052-019-0641-y 3103603910.1186/s 13052-019-0641-y PMC 6489191 · doi ↗ · pubmed ↗
- 6Derivation and external validation of the PLASMIC score Lancet Haematol Bendapudi PK Hurwitz S Fry A 1571644201710.1016/S 2352-3026(17)30026-128259520 · doi ↗ · pubmed ↗
- 7Thrombotic microangiopathies N Engl J Med Moake JL 58960034720021219202010.1056/NEJ Mra 020528 · doi ↗ · pubmed ↗
- 8Neurological manifestations of systemic lupus erythematosus and thrombotic thrombocytopenic purpura Stroke Petz LD 71972281977 https://doi.org/10.1161/01.STR.8.6.71956311610.1161/01.str.8.6.719 · doi ↗ · pubmed ↗