Case Report: Incidental late-onset Pompe disease diagnosis in a man with no clinical and instrumental evidence of neuromuscular dysfunction
Monica Sciacco, Sabrina Lucchiari, Letizia Bertolasi, Giacomo Pietro Comi, Stefania Corti, Dario Ronchi

TL;DR
A 33-year-old man was found to have late-onset Pompe disease through genetic screening, despite showing no symptoms or signs of the condition.
Contribution
This case highlights the possibility of incidental Pompe disease diagnosis in asymptomatic individuals through genetic screening.
Findings
The patient was homozygous for the GAA variant IVS1-32-13T>G, common in late-onset Pompe disease.
Most individuals with this variant in homozygosis remain asymptomatic, though some may develop symptoms over time.
Residual enzyme activity and protein levels do not correlate with clinical severity in these patients.
Abstract
Glycogen storage disease II or Pompe disease (PD), is a rare autosomal recessive disorder due to biallelic pathogenic variants in GAA, resulting in the enzymatic deficiency of alpha-1,4-glucosidase. Two clinical forms are recognized, namely, early onset (EOPD) and late-onset (LOPD). We present the case of an asymptomatic 33-year-old man who underwent a genetic screening for autosomal recessive disorders (parental prenatal counselling) and was found to carry the homozygous pathogenic GAA substitution NM_000152.5(GAA):c.-32-13T>G (IVS1). Neurological examination, serum CK levels, electromyography, muscle MRI, respiratory and cardiac screening were reported normal. We investigated the effects of the variant at transcript and protein levels in available tissues from the proband and his parents. The IVS1-32-13T>G variant (dbSNP: rs386834236, Clin Var ID: 4,027) occurs in 90% of Caucasian…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
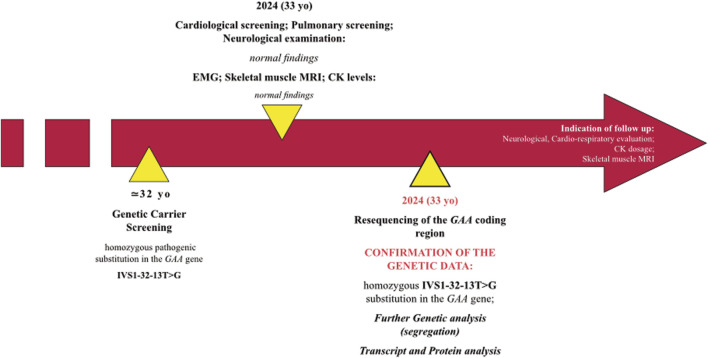 Figure 1
Figure 1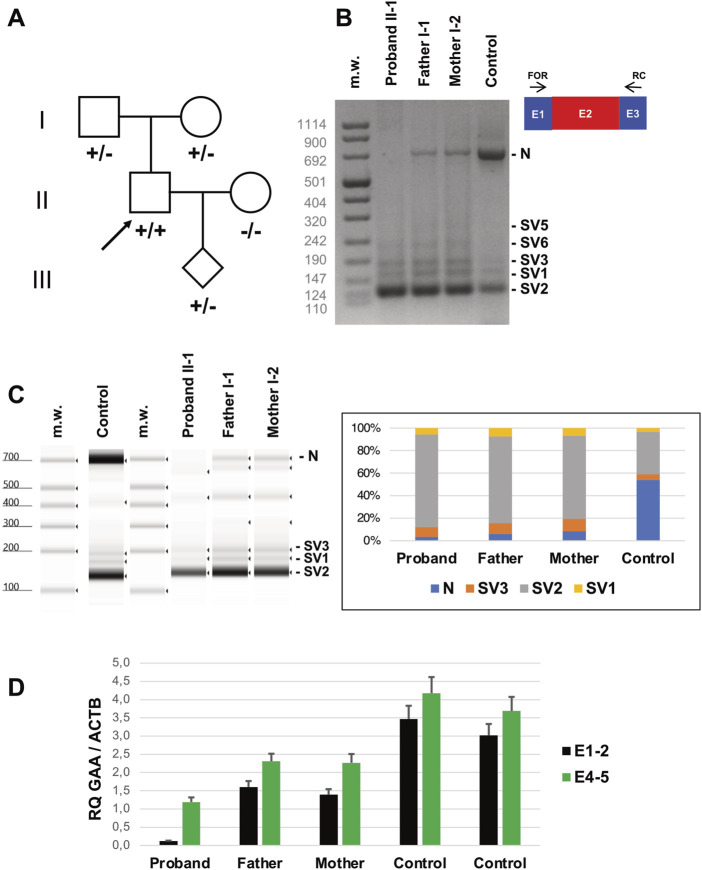 Figure 2
Figure 2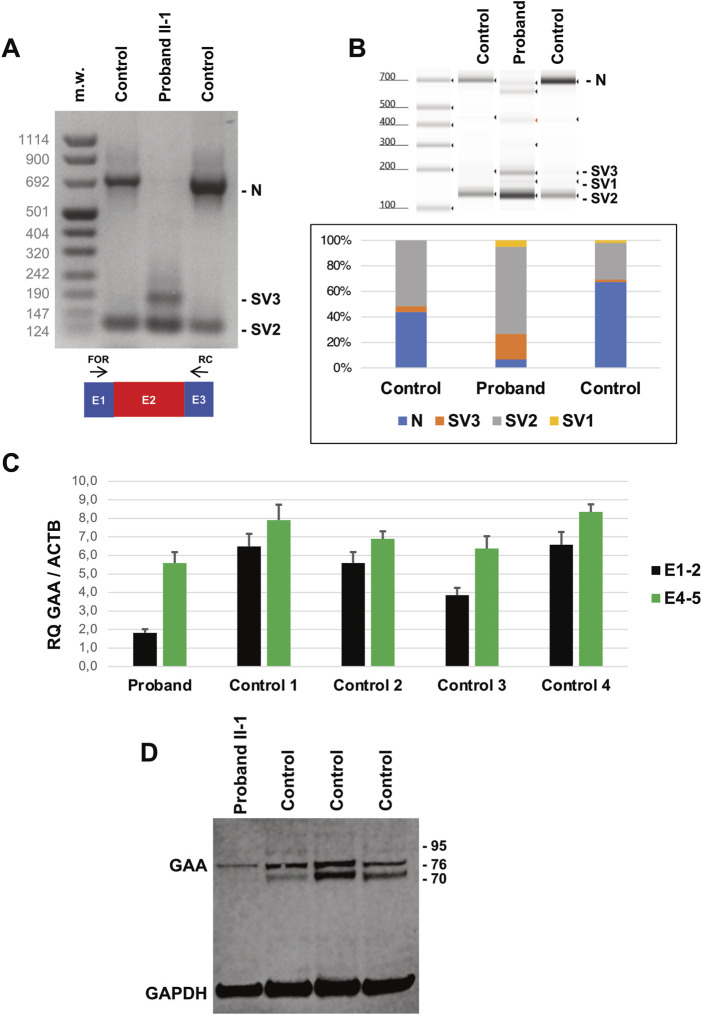 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLysosomal Storage Disorders Research · Glycogen Storage Diseases and Myoclonus · Trypanosoma species research and implications