Chromosome-level genome assembly of Ceroplastes pseudoceriferus Green, 1935 (Hemiptera: Coccidae)
Yaoguang Qin, Zhennan Wang, Jiangni Li

TL;DR
This paper presents the first high-quality chromosome-level genome assembly of a soft scale insect, which could help in managing these invasive pests.
Contribution
The study provides the first chromosome-level genome assembly for the Coccidae family.
Findings
The genome of Ceroplastes pseudoceriferus is 364.14 Mb with high contiguity and anchored into 18 chromosomes.
Approximately 54% of the genome consists of repetitive elements.
10,475 protein-coding genes were predicted, with 90.72% functionally annotated.
Abstract
Soft scales (Hemiptera: Coccidae) are significant polyphagous pests and majority of which are invasive species. The 364.14 Mb chromosome-level genome of Ceroplastes pseudoceriferus was assembled in this work, with a contig N50 length of 6.16 Mb and scafold N50 length of 21.24 Mb. Approximately 99.89% of assembled sequences were anchored into 18 chromosomes with the assistance of Hi-C reads. Furthermore, approximately 53.98% of the genome was composed of repetitive elements. In total, 10,475 protein-coding genes were predicted, of which 9503 (90.72%) genes were functionally annotated. The BUSCO analysis demonstrated the completeness of the genome annotation is 92.54%. This genome represents first high-quality chromosome level assembly of Coccidae, thereby advancing our knowledge of Coccidae insects and developing effective management strategies that protect crops, forests, and natural…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
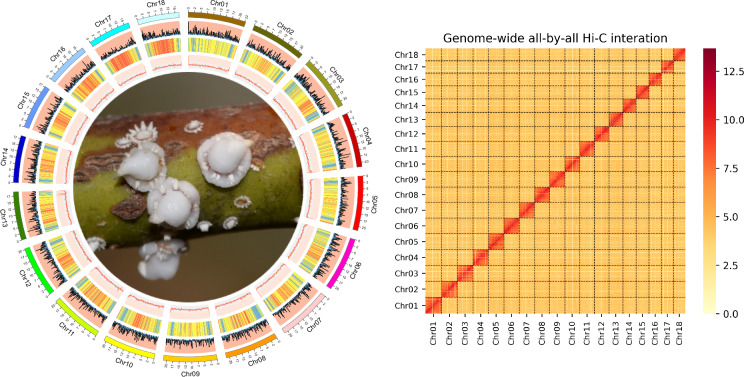 Figure 1
Figure 1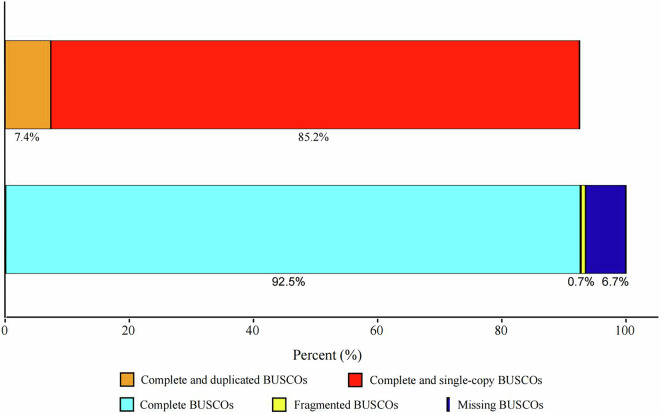 Figure 2
Figure 2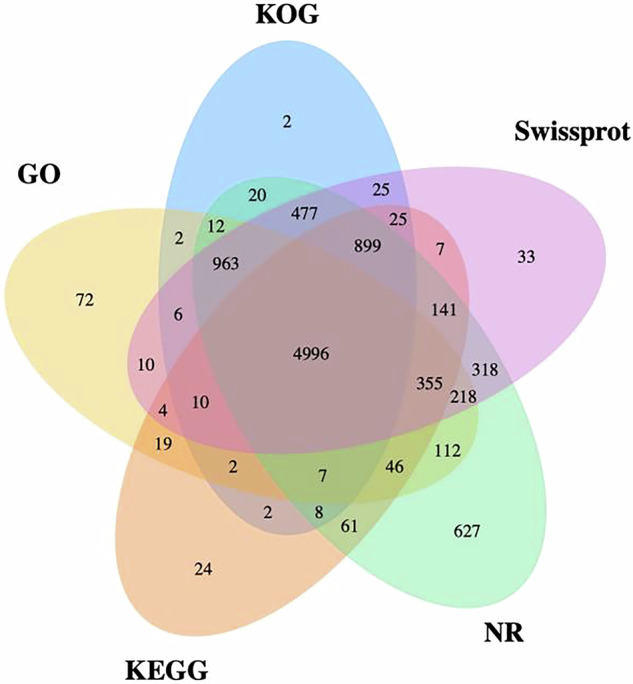 Figure 3
Figure 3- —Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences (L2024008) China Academy of Chinese Medical Sciences (Z2024032)
- —National Natural Science Foundation of China (32100363)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Research on scale insects · Insect symbiosis and bacterial influences
Background & Summary
Soft scales (Coccidae) is the third largest family within Subfamily Coccoidea behind Diaspididae (armoured scales) and Pseudococcidae (mealybugs)^1–3^. The majority of Coccidae species constitute significant polyphagous pests, posing a threat to economically vital agricultural and horticultural crops, as well as ornamental plants^4^. The inherent polyphagous characteristics and broad host plants of Coccidae species facilitate their widespread invasion globally. Furthermore, Coccidae insects play a pivotal role in ecosystem dynamics through complex interaction with predatory insects, parasitoids and entomopathogens^5^. Understanding the biology, behavior, and ecology of Coccidae insects is crucial for developing effective management strategies that protect crops, forests, and natural ecosystems. Until now, six genomes of Coccidae species are available from GenBank (Table 1), and only one scaffold-level genome has been published as research paper^6^. The scarcity of genomic resources poses a significant obstacle to intensive research endeavors aimed at advancing our knowledge of Coccidae insects.Table 1. Assembly features for genomes of Ceroplastes pseudoceriferus and other Coccidae species.Organism NameAssembly Stats Total Sequence LengthAssembly LevelAssembly Stats Total Number of ChromosomesAssembly Stats Contig N50Assembly Stats Scaffold N50Assembly Stats Number of ScaffoldsCeroplastes pseudoceriferus364,527,212Chromosome186,164,61221,238,50278Coccus hesperidum405,400,678Chromosome78,466,22954,289,48541Toumeyella liriodendri536,180,826Chromosome171,303,60630,207,406366Ericerus pela654,859,808Scaffold410,2341,243,5482396Coccus hesperidum484,566,906Scaffold399,49644,350,6616006Parthenolecanium corni231,203,063Contig14,265,238Ericerus pela660,374,735Contig660,240
Ceroplastes pseudoceriferus, first described by Green in 1935, is a highly polyphagous species of Coccidae. This species could infest a diverse array of plant species, encompassing 91 genera belonging to 56 plant families^1^. This broad host range underscores potential of C. pseudoceriferus as an invasive species. A high-quality reference genome of C. pseudoceriferus would be invaluable in enhancing our comprehension of ecological implications of Coccidae and facilitating the development of effective management strategies. In this study, we generated 35.11 Gb of MGI short-read sequencing and 102.21 Gb of Oxford Nanopore Technologies long-read sequencing, 77.20 Gb of high-throughput chromosome conformation capture (Hi-C). The final genome assembly size was 364.52 Mb with an N50 of 6.25 Mb. The karyotype of Ceroplastes pseudoceriferus is characterized by 18 chromosomes at the chromosome level. This conclusion is directly supported by the Hi-C scaffolding results in the genome assembly, where 99.89% of the assembled sequences (364.14 Mb) were anchored into 18 distinct chromosomes with an N50 of 21.24 Mb (Fig. 1).Fig. 1. Left: Overview of assembled C. pseudoceriferus genome.The outer layer of coloured blocks is a circular representation of the 18 chromosomes and circos demonstration of gene count (histogram), repeat density (heatmap) and GC content (line) from the outer to the inner circle, respectively. Right: The heatmap represents 18 chromosomes of the C. pseudoceriferus genome.
The assembly achieved an L50 value of 9, indicating that 50% of the total genome length (excluding ambiguous bases) is contained within the 9 longest scaffolds. This demonstrates high continuity in the assembly. The calculated auN value of 20,385,303.53 bp reflects a robust representation of long contiguous sequences, suggesting that the majority of the genome is assembled into large, uninterrupted segments. A total of 60 gaps were identified in the assembly, corresponding to regions of undetermined sequence (represented by runs of ‘N’ bases). This low gap count underscores the assembly’s high completeness. The assembly contains 6,000 ambiguous bases (N), accounting for <0.01% of the total genome length. Such minimal unresolved regions further validate the assembly’s reliability for downstream analyses.
We totally identified 998,369 repeat elements and the total length is 196,755,237 bp, accounting for 53.98% of whole sequence (Table 2). The total length of 23,595 tandem repeats (TR) is 1,374,491 bp, of which length of 10,466 simple sequence repeats (SSRs) is 123,925 bp. The annotation of transposable elements (TE) identified 873,421 repeat elements and the length is 179,535,462 bp, comprising 49.25% of the total. The annotation of non-coding RNAs (ncRNAs) totally resulted in 31 rRNA, 75 small RNA, 109 regulatory and 204 tRNA (Table 3). The gene prediction yielded a total of 10,475 genes, with an average gene length of 20,039 bp, an average CDS length of 1,701 bp, an average exons number per gene at 9, an average exon length of 199 bp, and an average intron length of 2,431 bp (Table 4).Table 2. Repetitive elements sequence statistics of the assembled genome.TypeNumber of elementsLength of sequence (bp)Percentage of sequence (%)TEsClass ILINEL24,919746,2130.20Unknown43,0187,569,9992.08I-Jockey3,377529,7660.15RTE-RTE3,253859,1320.24R11,414733,0180.20Other4,310459,2570.13LTRUnknown42,6297,267,2931.99Gypsy8,3291,639,8110.45Pao3,0071,311,5680.36Copia7,4802,085,6590.57Other3,372385,3070.11SINEUnknown8,093666,1560.18tRNA-Core-RTE1,795429,8430.12Other2,191621,8330.17Class IIDNAKolobok-T25,320428,7340.12TcMar-Tc15,396982,7300.27Unknown571,953118,603,00232.54Maverick6,4701,382,5540.38hAT-Ac29,6504,379,7261.20Crypton-I6,606949,4250.26CMC-Chapaev-31,962813,4200.22CMC-EnSpm2,6022,788,8890.77TcMar-Fot11,893514,5230.14Academ-11,404469,1090.13TcMar-Mariner6,4151,991,6630.55Other19,5412,482,8970.68MITEUnknown57,95114,793,8254.06RCHelitron19,0713,650,1101.00Total TEs873,421179,535,46249.25Tandem Repeatstandem_repeat13,1291,250,5660.34SSR10,466123,9250.03Total23,5951,374,4910.38Simple repeats2,243340,9960.09Unknown97,24615,186,4124.17Other1,759303,4520.08Low complexity10514,4240.00Total Repeats998,369196,755,23753.98Table 3Statistics of annotated non-coding RNAs.TypeCopy NumberAverage Length(bp)Total Length(bp)Percentage of sequence(%)rRNA (31)18S62,669.1716,0150.004428S66,343.0038,0580.01045.8S61589480.00035S13116.851,5190.0004small RNA (75)snRNA19113.112,1490.0006miRNA2380.651,8550.0005spliceosomal25140.123,5030.001other82471,9760.0005Regulatorycis-regulatory elements10948.615,2980.0015tRNAtRNA20475.3515,3720.0042Table 4Statistical results of gene structure prediction.Gene setTotal number of genesAverage gene length(bp)Average CDS length(bp)Average exons number per geneAverage exon length(bp)Average intron length(bp)TranscriptomeNGS RNA seq15,96430,114.733,481.199.79355.633,030.40PASA15,63729,593.233,477.109.75356.72,985.39HomoloyC. lectularius15,79552,434.271,417.847.72183.627,589.76A. pisum25,11835,660.831,328.275.84227.477,094.62N. lugens27,00138,850.771,323.465.67233.358,033.07H. halys20,82444,447.921,351.536.63203.797,651.91D. melanogaster17,76428,949.251,156.095.13225.376,729.98GeMoMa11,35329,039.921,443.027.13202.334,500.38De novoAUGUSTUS12,12419,075.141,665.978.42197.752,344.77FinalEVM10,47520,038.571,700.518.54199.032,430.89
The completeness of the predicted genes was assessed using Benchmarking Universal Single-Copy Orthologs (BUSCO)^7^, resulting in a high score of 92.54% (n = 1,265) (Fig. 2). This encompassed 85.15% (1164) single-copy, 7.39% (101) duplicated, 0.73% (10) fragmented, and 6.73% (92) missing BUSCOs, indicating a high integrity of gene prediction. Functional annotation indicated that a total of 90.72% (9503) of genes were annotated to five public databases including SwissProt^8^, the NCBI non-reduntant protein database (NR), Kyoto Encyclopedia of Gene and Genomes (KEGG)^9^, Eukaryotic Orthologous Groups of protein (KOG)^10^ and Gene Ontology (GO)^11^ (Fig. 3).Fig. 2BUSCO evaluation on the genome assembly completeness of the assembled genome.Fig. 3. Venn diagram of function annotations from various databases. The Venn diagram displays the overlap and uniqueness of functional gene annotations derived from five databases: SwissProt, Non-Reduntant Protein Database (NR), Kyoto Encyclopedia of Gene and Genomes (KEGG), Eukaryotic Orthologous Groups of protein (KOG) and Gene Ontology (GO).
Method
Sample collection, library construction and sequencing
First-instar larva of C. pseudoceriferus were collected from a single Camphor tree in Kunming, Yunnan Province, China (25°02′11″N, 102°42′31″E). Specimens were flash-frozen in liquid nitrogen and stored at −80 °C. Thousands of whole-body samples were used for DNA and RNA extraction to ensure sufficient biological material for multi-omics sequencing, including MGI short-read sequencing, Nanopore long-read sequencing, Hi-C sequencing and RNA sequencing. Genomic DNA (gDNA) was extracted using a QIAGEN® Genomic DNA Kit (Cat#13323). Total RNA was extracted using TRIzol® (TIANGEN) with DNase I treatment. All DNA and RNA extractions were conducted by Grandomics Biosciences (Wuhan, China) following manufacturer recommended protocols, meeting the quality specifications for library preparation and sequencing requirements.
For MGI sequencing, gDNA was randomly fragmented and libraries were prepared using standard protocols. After quality checks, the libraries were sequenced on the MGI DNBSEQ-T7RS platform, producing 35.11 Gb of raw data with 100 × average coverage.
For Nanopore sequencing^12^, size-selected DNA fragments were processed using the PippinHT system (Sage Science). Following end-repair and adapter ligation (SQK-LSK114 kit), library concentrations were quantified with a Qubit fluorometer. The prepared library was sequenced on a PromethION platform (Oxford Nanopore Technologies), yielding 102.21 Gb of long-read data at 287 × coverage for genome assembly.
For Hi-C sequencing, whole body samples were formaldehyde-fixed (2%) to crosslink DNA-protein complexes. Libraries were constructed through chromatin digestion, proximity ligation, and biotin pulldown following the protocol^13^. Sequencing on the MGISEQ-2000 platform generated 77.20 Gb of 150 bp paired-end reads (217 × coverage), enabling 3D chromatin architecture analysis.
For RNA sequencing, the Poly A mRNA was enriched via Dynabeads, fragmented, and reverse-transcribed using MGIEasy RNA Library Prep Kit V3.1 (Cat# 1000005276, MGI). Libraries were circularized and sequenced on DNBSEQ-T7RS, generating 13.45 Gb data for gene annotation.
Genome assembly
The raw Nanopore reads underwent de novo genome assembly using NextDenovo (v2.5.2)^14^ with default parameters. Four iterative correction rounds of Nextpolish (v1.2.4)^15^ polishing were subsequently applied using MGI short reads to enhance base accuracy. Assembly quality was verified through mapping all MGI reads to the assembled genome using BWA (Burrows-Wheeler Aligner, v0.7.12-r1039)^16^ and coverage assessment using Winnowmap2^17^ with parameters of “-x map-ont”. Base-level accuracy was calculated using samtools (v1.4)^18^ and BCFtools (v1.8.0)^19^ with default parameters. Mitochondrial sequence exclusion was performed via BLAST (v2.9)^20^ alignment against the NT database, followed by contaminant removal.
Chromosome anchoring
The 370 million paired-end reads underwent quality control through the standardized Hi-C data processing workflow^21^. Low-quality sequences (quality scores <20), adaptor sequences and sequences shorter than 30 bp were filtered out using fastp (v0.21.0)^22^. Clean reads were mapped using bowtie2 (v2.3.2)^23^ (-end-to-end–very-sensitive -L 30). HiC-Pro (v2.8.1)^21^ retained valid chromatin contacts while filtering invalid read pairs, including dangling-end, self-cycle, re-ligation, and dumped products. Scaffolds were organized into chromosomes using LACHESIS^24^, with key parameters CLUSTER_MIN_RE_SITES = 100, CLUSTER_MAX_LINK_DENSITY = 2.5, CLUSTER NONINFORMATIVE RATIO = 1.4, ORDER MIN N RES IN TRUNK = 60, ORDER MIN N RES IN SHREDS = 60. Final chromosomal orientations were manually verified and corrected based on chromatin interaction patterns.
Repeat annotation
Tandem repeats were systematically annotated through complementary approaches: simple sequence repeats (SSRs) were detected using GMATA (v2.2)^25^ with default parameters, while genome-wide tandem repeats were identified via Tandem Repeats Finder (TRF V4.07b)^26^ using sensitivity thresholds of “2 7 7 80 10 50 500 -f -d -h -r”. Transposable elements (TE) annotation was combinig ab initio and homology-based methods. ab initio repeat library was predicted using MITE-hunter^27^ with parameters of “-n 20 -P 0.2 -c 3” and RepeatModeler version open-2.0.4^28^ with parameters of “-engine wublast”, in which long terminal repeats (LTRs) were characterized using LTR_FINDER^29^, LTRharvest^30^ and LTR_retriever^31^. The obtained library underwent cross referencing against TEclass Repbase^32^ (http://www.girinst.org/repbase) for TE classification. Genome-wide TE masking was subsequently performed using RepeatMasker^33^ which simultaneously screened sequences against both the de novo library and RepBase reference. Overlapping TE annotations were resolved through BEDTools-based merging followed by manual curation to validate complex repeat architectures.
Annotation of non-coding RNAs (ncRNAs)
Non-coding RNA identification employed complementary database alignment and model-based prediction approaches. Transfer RNA genes were systematically predicted through tRNAscan-SE (v2.0)^34^ using eukaryotic configuration parameters. For comprehensive detection of small regulatory RNAs (miRNAs, snRNAs, snoRNAs) and ribosomal RNAs, a dual-strategy framework was implemented: Infernal (v1.1.2)^35^ facilitated covariance model searches against the Rfam Database^36^, while RNAmmer (v1.2)^37^ provided specialized prediction of rRNA subunits through hidden Markov model profiling. This integrated annotation pipeline ensured cross-validated identification of non-coding RNA elements across multiple functional categories.
Gene prediction
Gene annotation integrated three complementary methodologies applied to the repeat-masked genome. Homology-based predictions were generated through GeMoMa (v1.6.1)^38^, which mapped conserved protein sequences from five phylogenetically relevant species (Acyrthosiphon pisum^39^, Cimex lectularius^40^, Drosophila melanogaster^41^, Halyomorpha halys^42^ and Nilaparvata lugens^43^) onto the assembly. Concurrently, transcriptomic evidence was incorporated through RNA-seq alignment using STAR-2.7.3a^44^, followed by transcript assembly with StringTie2^45^ and ORF prediction via PASA (v2.3.3)^46^. For the ab initio prediction, RNA-seq-derived training sets processed through StringTie and PASA informed Augustus (v3.3.1)^47^ predictions. The three prediction streams were reconciled through EVidenceModeler (EVM)^46^, creating a unified gene set subsequently refined through TransposonPSI^48^ filtration to remove transposon associated genes. Structural annotation completeness was enhanced through PASA-based identification of untranslated regions (UTRs) and alternative splicing variants, with canonical isoforms selected based on maximum ORF length. Final gene models retained only protein-coding sequences validated through evolutionary conservation, transcriptional evidence, and ab initio support, ensuring both structural accuracy and biological relevance.
Functional annotation of gene
Gene functional characterization was conducted through systematic interrogation of five biological databases using complementary bioinformatics approaches. Protein domain architecture and Gene Ontology (GO) term assignments were determined via InterProScan^49^ analysis with default parameterization. Parallel BLASTp searches (E-value threshold ≤ 1e–5) against four major databases - SwissProt (manually curated proteins), NR (non-redundant sequences), KEGG (metabolic pathways), and KOG (eukaryotic ortholog clusters) - were performed on the EvidenceModeler-derived proteome, retaining only top-scoring alignments for each query sequence. The consensus functional annotations derived from these five independent evidence streams (InterProScan domain predictions and four BLAST-based database matches) were subsequently integrated through hierarchical evidence weighting, prioritizing experimentally validated annotations from SwissProt while incorporating complementary functional predictions from other resources. This multi-tiered annotation framework ensured comprehensive functional insights spanning molecular interactions, biological processes, and evolutionary relationships.
Data Records
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive^50^ in National Genomics Data Center^51^, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA019415^52^) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa. The whole genome sequence data and the annotation files for genome, ncRNA and repeat content are available from the Figshare repository^53–56^. The whole genome sequence are also deposited in the National Center for Biotechnology Information (NCBI) GenBank, with accession number GCA_050872025.1^57^, under BioProject PRJNA1273070.
Technical Validation
High mapping quality were confirmed in our results. After assembling ONT reads, BWA was used to align MGI short-reads with the assembled genome, resulted in mapping rate of 98.70%. The coverage of the genome by MGI short-reads is 99.72%. Winnowmap2 was used to align ONT reads with the assembled genome, resulted in mapping rate of 98.65%, and achieving a genome coverage of 99.99%. The combination of parthenogenetic tendencies in Coccidae, single-host sampling, and high assembly quality strongly suggests that these first-instar larvae had low genetic diversity, making them suitable for producing a coherent reference genome.