A Phenotypic Study of CRB1 Retinopathy Secondary to the Variant p.(Pro836Thr) Prevalent in Those of Black African Ancestry
Wendy M. Wong, Anthony G. Robson, Rebecca A. Baker, Gavin Arno, Joseph Van Aerschot, Siying Lin, Mariya Moosajee, Michel Michaelides, Omar A. Mahroo, Andrew R. Webster

TL;DR
This study examines the effects of a specific CRB1 gene variant common in people of African descent, showing it causes retinal disease with less severe symptoms than other variants.
Contribution
The study provides a detailed clinical characterization of the CRB1 p.(Pro836Thr) variant prevalent in Black African populations.
Findings
The p.(Pro836Thr) variant is associated with retinal dysfunction affecting both rods and cones.
Symptoms often begin in childhood, with reduced central vision as the main presenting issue.
Electrophysiology reveals generalized retinal dysfunction despite localized imaging findings.
Abstract
To comprehensively characterize the clinical consequences of the CRB1 variant p.(Pro836Thr). In African populations, this variant has an allele frequency of 0.329% (gnomAD v4.1.0). This study was a retrospective case series of 14 patients from 11 families with molecularly confirmed CRB1-associated retinal dystrophy, each possessing at least one p.(Pro836Thr) variant. The age at onset of visual symptoms, best-corrected visual acuity, imaging findings, and quantitative electrophysiologic measurements of retinal function were analyzed. The p.(Pro836Thr) variant was homozygous in four families and compound heterozygous in seven families. The familial origins included Nigeria (n = 4), Ghana (n = 3), the Caribbean region (n = 2), and Uganda (n = 1). The median follow-up was 7 years (interquartile range, 3–16). Symptom onset was most common in childhood (eight patients, 57.1%). Reduced…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
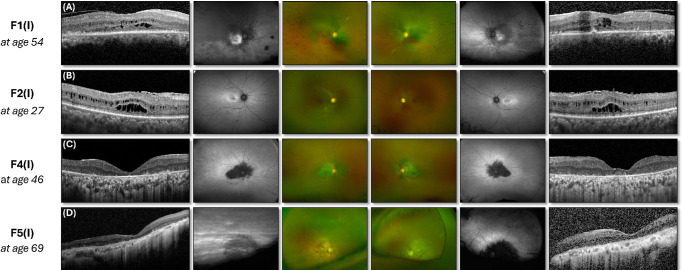 Figure 2
Figure 2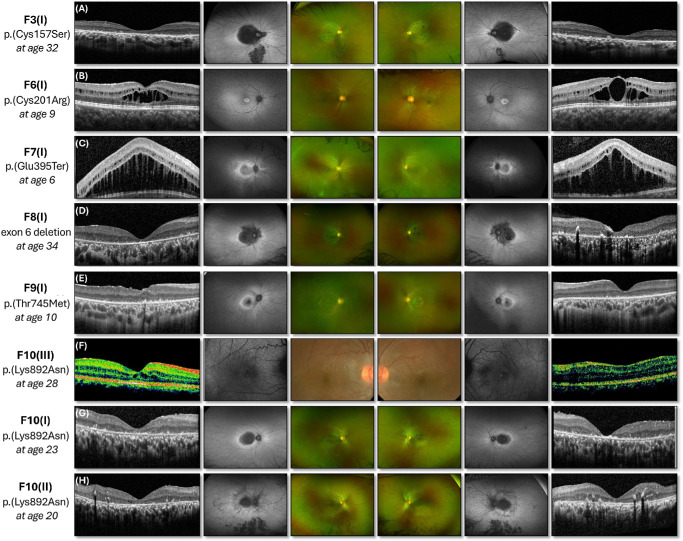 Figure 3
Figure 3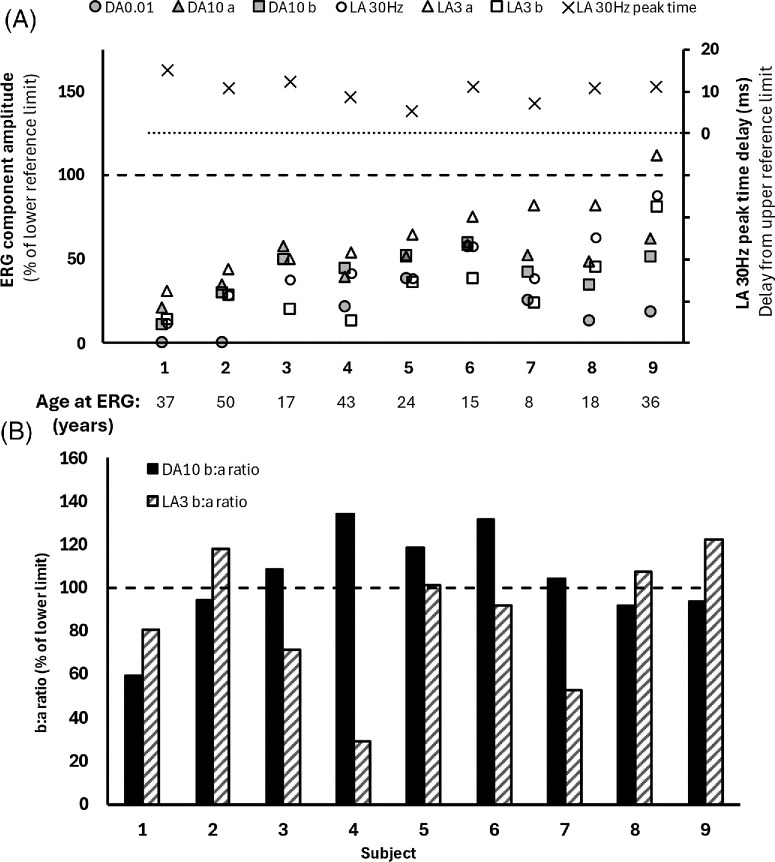 Figure 4
Figure 4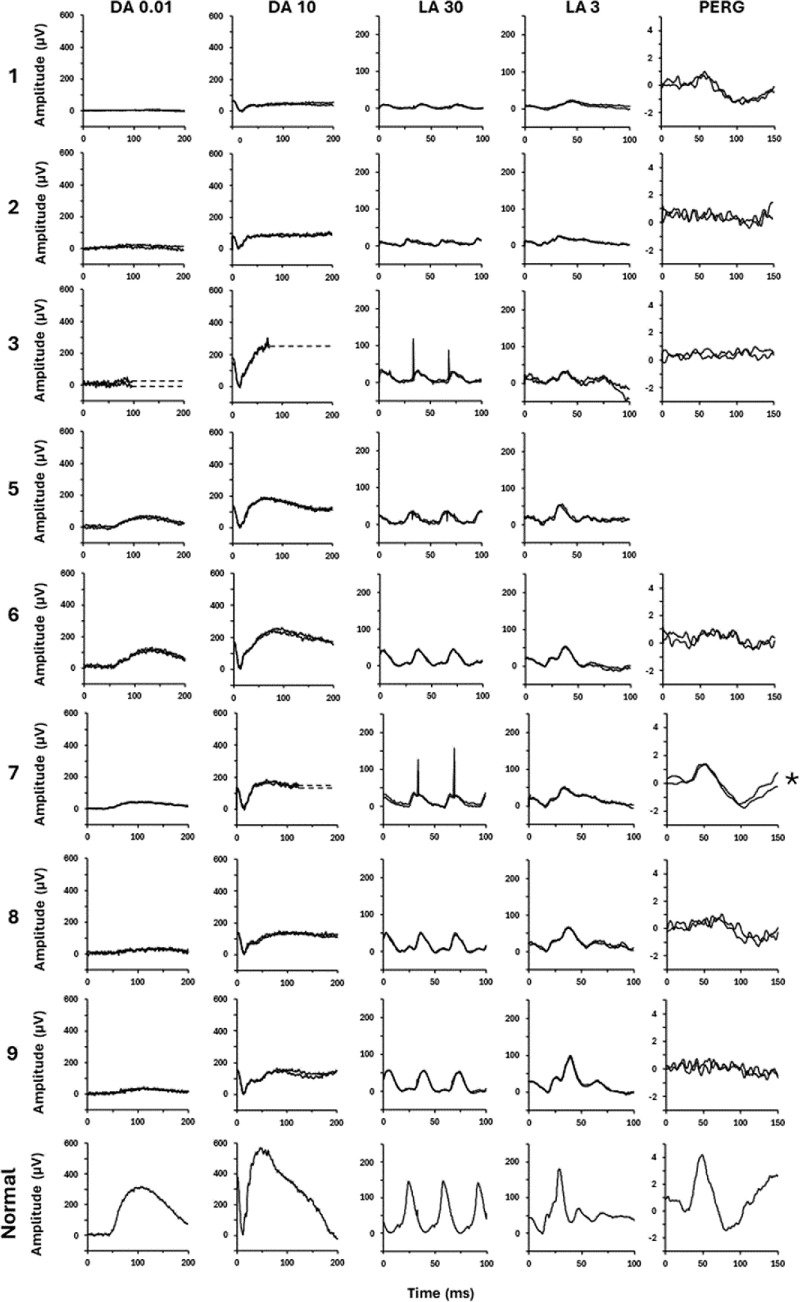 Figure 5
Figure 5| Allele 2 | At Presentation and at Final Review | Structural Imaging | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family(ID), Familial Origins, and Sex | Exon | Nucleotide and Protein Change | Presenting Symptoms | Age at Onset (y) | Age (y) | VA OD | VA OS | Affected Areas on FP and AF | Cystic Spaces on OCT | Initially Suspected Phenotype Clinically | Full-FieldERG Phenotype(Case No. According to |
| F1(I) | 7 | c.2506C>A | Decreased vision | Child-hood | 41 | 6/12 | 6/12 | Macula and peripheral retina | Present OU | Retinal dystrophy | Cone and rod dystrophy (case 1) |
| Ghana | p.(Pro836Thr) | 61 | 6/36 | 6/36 | |||||||
| Female | |||||||||||
| F1(II) | Decreased night vision | 43 | 51 | 6/12 | 6/18 | Not available | Present OU | Cone–rod dystrophy | Cone and rod dystrophy (case 2) | ||
| Ghana | then central vision | 53 | 6/12 | 6/12 | |||||||
| Male | |||||||||||
| F2(I) | 7 | c.2506C>A | Decreased vision | 21 | 27 | 6/12 | 6/9 | Macula only | Present OU | Macular dystrophy | N/A |
| Nigeria | p.(Pro836Thr) | 30 | 6/12 | 6/12 | |||||||
| Female | |||||||||||
| F3(I) | 2 | c.470G>C | Decreased | Child-hood | 6 | 6/36 | 6/36 | Macula and nasal to OD | Absent | Cone–rod dystrophy | Cone and rod dystrophy (case 3) |
| St Lucia and France | p.(Cys157Ser) | vision | 34 | 6/60 | 6/60 | ||||||
| Male | |||||||||||
| F4(I) | 7 | c.2506C>A | Decreased vision | 40 | 43 | 3/60 | 6/12 | Macula and nasal to OD | Absent | Cone–rod dystrophy | Cone and rod dystrophy (case 4) |
| Nigeria | p.(Pro836Thr) | 46 | 1/60 | 6/18 | |||||||
| Female | |||||||||||
| F5(I) | 7 | c.2506C>A | Decreased central and night vision | 40s | 68 | CF | CF | Macula and nasal to OD | Absent | Cone–rod dystrophy | N/A |
| Jamaica | p.(Pro836Thr) | ||||||||||
| Male | |||||||||||
| F6(I) | 2 | c.601T>C | Decreased vision | 5 | 9 | 6/15 | 6/15 | Macula only | Present OU | Foveal schisis | N/A |
| Ghana | p.(Cys201Arg) | 12 | 6/12 | 6/12 | |||||||
| Male | |||||||||||
| F7(I) | 6 | c.1183G>T | Decreased vision | 6 | 7 | 6/48 | 6/48 | Macula and nasal to OD | `Present OU | Foveal schisis | Cone–rod dystrophy (pediatric protocol with skin electrodes) |
| Nigeria and Zimbabwe | p.(Glu395Ter) | 14 | 6/30 | 6/30 | |||||||
| Male | |||||||||||
| F8(I) | 6 | Deletion of exon 6 | Decreased vision | 23 | 23 | 6/60 | 6/60 | Macula and nasal to OD | Absent | Retinal dystrophy | Cone and rod dystrophy (case 5) |
| Ghana | 39 | CF | 6/60 | ||||||||
| Female | |||||||||||
| F9(I) | 7 | c.2234C>T | Decreased vision | 6 | 6 | 6/38 | 6/38 | Macula only | Absent | Macular dystrophy | Mild cone dystrophy (pediatric protocol with skin electrodes) |
| Unknown | p.(Thr745Met) | 13 | 6/38 | 6/48 | |||||||
| Male | |||||||||||
| F10(I) | 7 | c.2676G>C | Decreased vision | 15 | 15 | 6/15 | 6/15 | Macula only | Absent | Macular dystrophy | Cone and rod dystrophy (case 6) |
| Nigeria | p.(Lys892Asn) | 31 | 6/95 | 6/75 | |||||||
| Female | |||||||||||
| F10(II) | Decreased | 9 | 9 | 6/9 | 6/9 | Macula and peripheral retina | Absent | Retinal dystrophy | Cone and rod dystrophy (case 7) | ||
| Nigeria | vision | 24 | 6/60 | 6/36 | |||||||
| Female | |||||||||||
| F10(III) | Decreased vision | Child-hood | 18 | 6/12 | 6/18 | Macula only | Present OD | Retinal dystrophy | Rod–cone dystrophy (case 8) | ||
| Nigeria | 19 | 6/12 | 6/12 | ||||||||
| Male | |||||||||||
| F11(I) | 3 | c.716G>A | Decreased | 12 | 33 | 6/24 | 6/24 | Macula and peripheral retina | Present OU | Retinal dystrophy | Rod–cone dystrophy (case 9) |
| Uganda | p.(Cys239Tyr) | vision | 56 | 6/60 | 3/60 | ||||||
| Female | |||||||||||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRetinal Development and Disorders · Retinal and Macular Surgery · Drug-Induced Ocular Toxicity
Crumbs (Crb), first discovered in Drosophila, is an evolutionarily conserved protein that recruits other cell polarity regulators to form the Crb complex, which is essential for retinal development and regulation of cell adhesion.1^–^4 The Crumbs homolog 1 (CRB1) gene is expressed in the brain and the retina, where it localizes to the microvilli of Müller glial cells and inner segments of photoreceptors.5^,^6 Biallelic disease-causing variants in CRB1 (OMIM *60420) are associated with a broad spectrum of inherited retinal disease (IRD) phenotypes: (1) Leber congenital amaurosis (LCA); (2) early-onset severe retinal dystrophy; (3) rod–cone dystrophy, which can occur in conjunction with preserved para-arteriolar retinal pigment epithelium (PPRPE) and/or a Coats-like exudative vasculopathy; (4) cone–rod dystrophy; (5) macular dystrophy; (6) foveal retinoschisis; and (7) fenestrated slit maculopathy.7^–^12
The CRB1 gene spans 210 kb of genomic DNA on chromosome 1q31.3 and consists of 12 exons.6^,^13 Thus far, mRNA corresponding to three CRB1 transcripts has been identified in the retina.6^,^14 The canonical 1406-aa isoform (CRB1-A, GenBank accession number MT470365) is most abundant during development; it has a large extracellular domain containing multiple epidermal growth factor–like domains and three laminin A globular (AG)-like domains (Fig. 1), as well as a transmembrane domain and highly conserved 37-aa intracellular domain.6^,^9^,^15 A second validated transcript is predominant in the adult retina and encodes a shorter 1003-aa protein (CRB1-B, GenBank accession number MT470366).15^,^16 The third validated transcript encodes a 754-aa isoform (CRB1-C, GenBank accession number MT470367) without transmembrane and intracellular domains.6^,^15 More than 450 variants have been linked to CRB1-associated retinal dystrophies, with the majority clustering in exons 7 (27%) and 9 (41%), which encode the second and the third laminin AG-like domains.11^,^16
A growing number of molecularly diagnosed IRD cohorts have revealed that several CRB1 variants have a relatively high prevalence in specific ethnic populations.11^,^17^,^18 For example, the in-frame deletion p.(Ile167_Gly169del) has an overall population allele frequency of 0.123%, which rises to 0.156% in European populations (gnomAD v4.1.0).17 In a Chinese cohort, the p.(Gly1226Ter) variant is especially prevalent.18 In African populations the p.(Pro836Thr) variant has a high allele frequency of 0.329% (gnomAD v4.1.0), and was recently recognized as a cause of CRB1 retinopathy in four families.19 This study aims to comprehensively characterize the clinical consequence of the CRB1 variant p.(Pro836Thr).
Methods
This retrospective study was approved by the local ethics committee (Moorfields Eye Hospital and the Northwest London Research Ethics Committee) and adhered to the tenets of the Declaration of Helsinki. Patients were identified using an in-house database (OpenEyes, London, UK) and only included patients with molecularly confirmed CRB1-associated retinal dystrophy with at least one (NM_201253.3) c.2506C>A; p.(Pro836Thr) variant. The molecular diagnosis was established using next-generation sequencing (panel testing of retinal dystrophy genes, whole exome sequencing, or whole genome sequencing). The sequence variants were interpreted according to the guidelines from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.20 Population allele frequencies were derived from gnomAD v4.1.0.
The phenotype was determined through a comprehensive review of the clinical history, multimodal imaging and available functional assessments from electrophysiological testing. Relevant clinical information extracted from the electronic medical records included: (1) demographics, (2) genetic information, (3) age at onset, (4) presenting symptoms, (5) family history, and (6) best-corrected Snellen visual acuity at the time of presentation. In patients reviewed from 2014 onward, the Optos confocal scanning laser ophthalmoscopy system (Optos, Dunfermline, UK) was used to acquire ultra-widefield (UWF) pseudocolor fundus photographs and UWF green wavelength autofluorescence; the SPECTRALIS system (Heidelberg Engineering, Heidelberg, Germany) was used to acquire spectral-domain optical coherence tomography (OCT) scans.
Pattern electroretinography (PERG) and full-field ERG were performed to incorporate the International Society for Clinical Electrophysiology of Vision (ISCEV) standards in nine individuals (age range, 8–50 years), using gold foil corneal recording electrodes.21^,^22 The two youngest (both 5 years old) were tested using skin electrodes according to abbreviated protocols, including one without mydriasis tested using non-Ganzfeld flash stimulation.23 PERG P50 was used as a measure of macular function, and the full-field ERG was used to assess generalized (mainly peripheral) rod and cone system function. The main components of the ISCEV dark-adapted (DA) and light-adapted (LA) ERGs were quantified and compared with age-matched control data from healthy subjects (age range, 10–79 years), with ERG amplitudes plotted as a percentage of the age-matched lower limit of the reference range and peak times plotted as a difference from the age-matched upper limit of the reference range.24^,^25
Results
Fourteen individuals (seven females, seven males) from 11 families were identified. The familial origins included Nigeria (four pedigrees), Ghana (three pedigrees), the Caribbean region (two pedigrees), and Uganda (one pedigree). The p.(Pro836Thr) variant was homozygous in four families (Table). In seven families that were compound heterozygous for the p.(Pro836Thr) variant, the second variant was a missense in five families, a nonsense variant in one family, and a deletion of exon 6 in one family. The amino acid residues predicted to be affected are illustrated in Figure 1.
Symptom onset was most commonly in mid- to late childhood (eight patients, 57.1%), although three patients did not observe symptoms until their fourth decade (21.4%). Reduced central vision was the most frequent presenting symptom (12 patients, 85%). In two patients, nyctalopia either preceded or coincided with the observation of reduced central vision. Nystagmus was present only in one patient, G15465(I). All patients had reduced visual acuity (range, 6/9 to counting fingers) and bilateral disease at presentation (Figs. 2, 3). The median follow-up was 7 years (interquartile range [IQR], 3–16). At the last follow-up, the median age was 31 years (IQR, 19–46), and deterioration in visual acuity was documented in nine patients (64.3%).
Multimodal imaging of patients homozygous for the p.(Pro836Thr) variant. (A–D) The optical coherence tomography scans of all patients show an abnormally laminated and thickened retina. Macular cystic spaces, when present, affected both the inner and outer nuclear layers (A, B).
Multimodal imaging of patients who are compound heterozygous for the p.(Pro836Thr) variant (A–H). Marked intrafamilial phenotypic variation between three siblings was observed in F11; the youngest sibling has the earliest symptom onset, and additional involvement of the peripheral retina was evident on widefield fundus imaging.
Nummular pigmentary changes in the macular region were visible in six patients on UWF fundus photography (magnified view of the central retina in Supplementary Fig. S1), with corresponding UWF autofluorescence demonstrating confluent hypo-autofluorescence involving the fovea, parafovea, and perifovea. The hypo-autofluorescence extended nasal to the optic disc in all but one case (Supplementary Fig. S1). Intraretinal migration of retinal pigment epithelium (RPE) cells was visible as intraretinal hyper-reflective foci on OCT and could be identified within both the outer and inner retinal layers (Figs. 3D, 3H). Mid-peripheral bone-spicule–like pigmentary deposits were observed in three patients (Figs. 2A, 3A, 3H), with PPRPE evident only in a single patient (Fig. 3H).
Electrophysiology testing was performed in 11 patients. The ISCEV standard full-field ERGs showed a high degree of interocular symmetry based on amplitudes of the DA 0.01, DA 3, and DA 10 ERG a-waves and b-waves; LA 30-Hz ERG and LA 3 ERG b-waves (slope = 0.95; r^2^ = 0.96); and on the peak times of the DA 3 and DA 10 ERG a-waves and b-waves and LA 30-Hz and LA 3 ERGs (slope = 1.05; r^2^ = 0.98).
The ISCEV full-field ERG findings are quantified in Figure 4, and examples of recordings are shown in Figure 5. The DA 0.01 and DA 10 ERG a-waves and b-waves were subnormal in nine of nine cases, and LA ERG components were subnormal in all but one individual, with a preserved LA 3 ERG b-wave (Fig. 4A, case 9). There was LA 30-Hz flicker delay in all nine cases (mean delay, 10 ms; range, 5–15 ms). The findings indicate generalized retinal dysfunction at the level of the photoreceptors, including two with a clear rod–cone pattern of dysfunction (cases 8 and 9) and others with a similar degree of rod and cone system involvement (cases 1–7). The LA 3 ERG b:a ratios were subnormal in five patients (cases 1, 3, 4, 6, and 7); the DA 10 ERG b:a ratios were additionally mildly subnormal in four patients (cases 1, 2, 8, and 9), including those with undetectable or severely reduced rod-system–specific DA 0.01 ERGs. DA 10 ERG b-waves showed delay in three patients (cases 6, 7, and 9). There was no obvious correlation between age and the severity of ERG abnormalities (Fig. 4A).
Full-field findings in subjects who were tested according to ISCEV standard methods. (A) The amplitudes of the DA 0.01 ERG, DA 10 ERG a-waves and b- waves, and LA 30-Hz ERG and LA 3 ERG a-waves and b- waves are plotted as a percentage of the age-matched lower limit of the reference (“normal”) range, with values arranged in ascending order of LA 3 ERG a-waves for clarity (primary y-axis). The LA 30-Hz peak times are plotted as a difference from the age-matched upper limit of the reference range (secondary y-axis). All electrophysiological recordings showed a high degree of interocular symmetry and are shown for right eyes only. (B) The DA 10 ERG and LA 3 ERG b:a ratios are compared with the lower limit of the reference range and are shown as a percentage.
Representative ERG recordings for cases 1 to 3 and 5 to 9, tested according to ISCEV standard methods and from a representative control subject (N) for comparison. Patient waveforms are superimposed to demonstrate reproducibility. Broken lines replace blink and eye movement artifacts for clarity. For case 7 (), a high-frequency electrical artifact was removed from the PERG by digital filtering, post-acquisition. Traces from case 4 are not shown (recorded with a different recording system).*
Six of nine cases had undetectable pattern ERG P50 components, consistent with severe macular involvement; P50 was detectable but subnormal in cases 1, 6, and 7, reduced by 55%, 65%, and 20%, respectively (Fig. 5).
Three individuals underwent follow-up recordings (Supplementary Fig. S2); two showed a high degree of ERG stability after 4 or 5 years, and one subject tested after 9 years had mildly increased LA 30-Hz peak times (by 3 ms), consistent with marginal worsening of cone system function. One of the two 5-year-old children tested with lower eyelid skin electrodes showed ERG evidence of a cone–rod dystrophy; the other showed mild 30-Hz flicker ERG delay, consistent with mild generalized cone system dysfunction. Undetectable pattern ERGs indicated macular involvement in both.
Discussion
This study of CRB1-related retinal dystrophy reports detailed phenotyping of patients molecularly confirmed to have the p.(Pro836Thr) variant and characterizes their key clinical and electrophysiological features. The study extends previous investigations of CRB1-related retinopathy,18 highlighting the association of the p.(Pro836Thr) variant with African ancestry. The allele frequency of this variant is markedly different among various genetic ancestry groups; although it is observed at 0.329% in Africans/African Americans, it is absent in Europeans and Asians (gnomAD v4.1.0). Of note, research on IRDs has been comprehensively performed in Europe and North America, but a substantial proportion of patients remain genetically undiagnosed in Asia and Africa, which represent over 60% of the global population.26^,^27 The underrepresentation of specific racial/ethnic groups in population databases and disease–gene association databases makes variant interpretation more challenging and can lead to both over- and underdiagnosis of an inherited disorder.28^,^29 It is reasonable to infer that many causative IRD variants remain unknown, and expanding our knowledge of ethnic-specific variants is important to enhance diagnostic precision and support equitable health care.
Biallelic pathogenic variants in the CRB1 gene are known to result in a diverse spectrum of retinopathies with phenotypic variability.30 Interestingly, although LCA is the most commonly reported phenotype for CRB1-associated retinal dystrophy (43%) and CRB1 is implicated in 7% to 17% of autosomal recessive LCA cases, none of the patients in our cohort exhibited this phenotype.6^,^31 Literature review for this specific variant identified an additional 15 patients from nine publications, six of whom were homozygous for this variant.9^–^12^,^16^,^32^–^36 Altogether, 28 patients have an established genotype–phenotype correlation, and only one homozygous patient has been reported to exhibit the LCA phenotype.33 The unusual severity of this reported patient raises considerations of a hemizygous deletion, which may occur more commonly than recognized by currently utilized molecular diagnostic techniques.37 Additionally, none of the patients in our cohort had developed a Coats-like exudative vasculopathy after a median follow-up of 7 years, with a median age of 31 years at the last follow-up. Our study suggests that the p.(Pro836Thr) tends to confer a phenotype less severe than LCA, as the symptom onset and presentation occurred in mid- to late childhood, and the majority of patients did not have nystagmus, in contrast to vision loss and nystagmus in early infancy characteristic of LCA.7
In our cohort, there was considerable phenotypic diversity even among patients homozygous for the p.(Pro836Thr) variant, and between siblings with identical CRB1 variants. The biological mechanisms driving this broad phenotypic variability are of scientific interest. An estimated 95% of multiexon genes generate multiple mRNA isoforms, and this could play an important role in modifying disease severity.38^,^39 CRB1 has three isoforms found to be expressed at meaningful levels in the retina: CRB1-A, CRB1-B, and CRB1-C.6^,^14 Analysis of RNA isolated from retinas of donor eyes has revealed that CRB1-B is the predominant isoform in adult retina*.* Compared to the canonical isoform CRB1-A, there are differences at its 5′ and 3′ ends: Distinct promoters at the 5′ exon drive CRB1-A expression in Müller glial cells and CRB1-B expression in photoreceptors, whereas the 3′ exons encode different intracellular domains that may interact with different intracellular partners.14 Although limited genotype–phenotype correlations have been found for CRB1-linked IRDs, other than the association of the in-frame deletion p.(Ile167_Gly169) with a restricted macular dystrophy phenotype, null variants are associated with greater disease severity, and this is perhaps explained by a disruption of all CRB1 isoforms.17^,^19 The p.(Pro836Thr) is predicted to affect all Müller cell and photoreceptor isoforms.16 This predicted effect is broadly consistent with the available electrophysiological findings in our patients showing generalized photoreceptor dysfunction, mostly with similar involvement of rods and cones, but with additional evidence of dysfunction post-phototransduction, or inner retinal, in six of nine cases. It was not possible to infer inner retinal dysfunction from the two patients with the most severe rod photoreceptor involvement (Fig. 4, cases 1 and 2), because in these two cases the reduced DA10 ERG b:a ratios may reflect the “photopic hill” phenomenon manifesting under conditions of dark adaptation, in the absence or near-absence of rod function, as has been described in some other conditions that selectively or predominantly affect the rod photoreceptors.40^–^42
In compound heterozygous patients, it remains to be determined how the second variants disrupt one or all isoforms of CRB1 and how the degree of functional preservation in each isoform and level/ratio of the isoforms could collectively modify the disease severity and progression. An additional potential disease modifier stems from the presence of the CRB1 gene paralog CRB2 (OMIM 609720), which also encodes multiple protein isoforms.2^,^6^,^43 CRB2 localizes to the (1) subapical region of Müller glial cells, (2) inner segment of photoreceptor inner segments, and (3) RPE cells.6^,^44^,^45 Another CRB1 paralog, CRB3, localizes to the Müller glial cells, photoreceptors, rod bipolar cells, and vascular pericytes, but variants in this gene have yet to be linked to retinal disease.4^,^46^,^47 CRB2 has been associated with autosomal recessive non-syndromic retinitis pigmentosa (RP).48 In mouse models, knockout of Crb1 was associated with an RP-like phenotype, but additional knockout of Crb2 gave rise to an LCA-like phenotype.46 Further, in Crb double-mutant mice, recombinant adeno-associated virus (rAAV)-mediated human CRB2 gene delivery to Müller cells was demonstrated to partially preserve retinal morphology.49 Notably, expressing human CRB1 in tissues lacking endogenous mouse Crb1 led to severe infiltration of immune cells, likely from a compromised blood–retinal barrier and immune privilege. However, expression of human CRB2 did not trigger a similar immune response and may relate to ubiquitous expression in extraocular tissues reducing its immunogenicity.47 These preclinical studies provide preliminary evidence that CRB2 gene therapy might have the potential to improve retinal function and morphology in *CRB1-*associated retinal dystrophy.47^,^49
In conclusion, the p.(Pro836Thr) variant is particularly prevalent in those of Black African ancestry. This variant is generally associated with a milder phenotype and is less likely to be associated with a severe LCA phenotype; however, electrophysiology in this series indicates that dysfunction is not restricted to the macula, as suggested in some but not all of the retinal imaging findings. Although the clinical variability remains incompletely understood, recent insights into the isoforms of CRB1 suggest that post-transcriptional processes within Müller glial cells and photoreceptors are potential modifiers of disease severity.16 More work is needed to understand whether co-occurrence of variants in the gene paralog CRB2 contributes to the mutation load and further disrupts retinal homeostasis.47 Further analysis of the CRB1 isoforms expressed in patient-derived retinal organoids would be helpful to unravel the relevant molecular mechanisms and their possible impact on DNA- or RNA-editing therapeutic approaches.
Supplementary Material
Supplement 1
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Tepass U . Crumbs, a component of the apical membrane, is required for zonula adherens formation in primary epithelia of Drosophila. Dev Biol. 1996; 177(1): 217–225.8660889 10.1006/dbio.1996.0157 · doi ↗ · pubmed ↗
- 2Bulgakova NA, Knust E. The Crumbs complex: from epithelial-cell polarity to retinal degeneration. J Cell Sci. 2009; 122(pt 15): 2587–2596.19625503 10.1242/jcs.023648 · doi ↗ · pubmed ↗
- 3Richard M, Roepman R, Aartsen WM, et al. Towards understanding CRUMBS function in retinal dystrophies. Hum Mol Genet. 2006; 15 Spec No 2: R 235–R 243.16987889 10.1093/hmg/ddl 195 · doi ↗ · pubmed ↗
- 4Quinn PMJ, Wijnholds J. Retinogenesis of the human fetal retina: an apical polarity perspective. Genes (Basel). 2019; 10(12): 987.31795518 10.3390/genes 10120987 PMC 6947654 · doi ↗ · pubmed ↗
- 5Wright GA, Rodriguez-Martinez AC, Conn H, et al. Enhanced learning and memory in patients with CRB 1 retinopathy. Genes (Basel). 2024; 15(6): 660.38927596 10.3390/genes 15060660 PMC 11203261 · doi ↗ · pubmed ↗
- 6Quinn PM, Pellissier LP, Wijnholds J. The CRB 1 complex: following the trail of crumbs to a feasible gene therapy strategy. Front Neurosci. 2017; 11: 175.28424578 10.3389/fnins.2017.00175 PMC 5380682 · doi ↗ · pubmed ↗
- 7Kumaran N, Moore AT, Weleber RG, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. 2017; 101(9): 1147–1154.28689169 10.1136/bjophthalmol-2016-309975 PMC 5574398 · doi ↗ · pubmed ↗
- 8Kousal B, Dudakova L, Gaillyova R, et al. Phenotypic features of CRB 1-associated early-onset severe retinal dystrophy and the different molecular approaches to identifying the disease-causing variants. Graefes Arch Clin Exp Ophthalmol. 2016; 254(9): 1833–1839.27113771 10.1007/s 00417-016-3358-2 · doi ↗ · pubmed ↗