Correction to “Ab Initio Design of Molecular Qubits with Electric Field Control”
William T. Morrillo, Herbert I. J. Cumming, Andrea Mattioni, Jakob K. Staab, Nicholas F. Chilton

Abstract
Click any figure to enlarge with its caption.
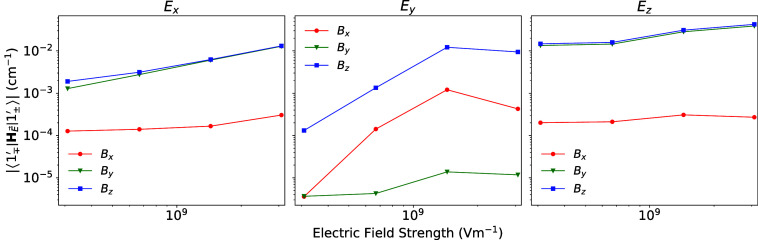 Figure 1
Figure 1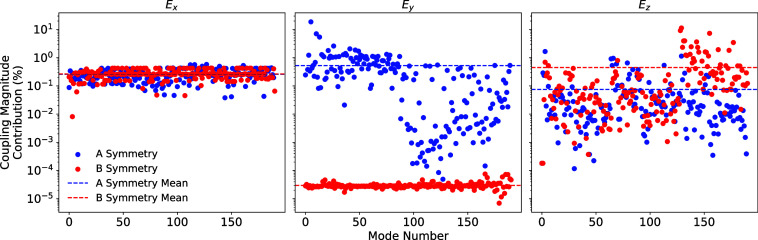 Figure 2
Figure 2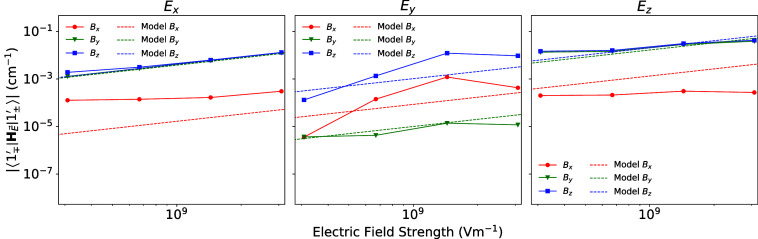 Figure 3
Figure 3- —Royal Society10.13039/501100000288
- —University of Manchester10.13039/501100000770
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMolecular Junctions and Nanostructures · Spectroscopy and Quantum Chemical Studies · Gold and Silver Nanoparticles Synthesis and Applications
Our work used computational methods to study control of electron spins using applied electric fields.? After we determine the molecular distortion due to an applied electric field, we approximate the electronic Hamiltonian using a linear vibronic coupling (LVC) model. The first step of this approximation is to map the distorted geometry onto the equilibrium geometry where the LVC model was parametrized in order to minimize the Euclidean distance between the molecular structures and hence minimize the truncation error of the LVC approximation. However, the implementation of the Kabsch algorithm? used to align molecular geometries was incorrect, giving an incorrect molecular rotation. Hence, the resulting crystal field parameters (B _ k _ ^ q ^) obtained from evaluating electric field distorted geometries using the LVC were inaccurate. The error in the crystal field parameters increases as the distortion of the molecule grows, resulting in large errors for geometries at high electric fields. The alignment error also resulted in an effective change of reference frame away from the molecular reference frame. In-turn, our magnetic fields were no longer oriented along the molecular axes thereby affecting the magnetic field orientation dependence of the spin-electric coupling in Figure 5. We also found a small error in the ab initio spin-electric coupling in Figures 2c and 5, where the spin-electric coupling was 1 order of magnitude too small due to being calculated using a magnetic field strength of 32 mT rather than 320 mT. In this correction Figure replaces Figure 2c in the original work, while Figures and ? replace Figures 3 and 5, respectively.
In the original work, we believed that the discrepancy in the spin-electric coupling between the electric field model and ab initio simulations when the electric field was oriented along the y-axis was attributable to the model’s inability to replicate the symmetry-preserving behavior we observed in our ab initio findings. However, with the corrected rotation here, the electric field model accurately predicts the magnetic field orientation dependence for all electric field orientations (Figure). Our conclusions surrounding the electric field model has not changed, and in fact, we now see improved agreement with the electric field model; it is more robust than we originally thought.
The data in the associated repository (doi: 10.48420/26617561) and the code implicated (spin_phonon_suite) have been updated.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Morrillo W. T.Cumming H. I. J.Mattioni A.Staab J. K.Chilton N. F.Ab Initio Design of Molecular Qubits with Electric Field Control J. Am. Chem. Soc.2024146258412585110.1021/jacs.4c 09109 PMC 1142102739237481 · doi ↗ · pubmed ↗
- 2Kabsch W.A solution for the best rotation to relate two sets of vectors Acta Crystallogr., Sect. A 19763292292310.1107/S 0567739476001873 · doi ↗