Detection of a new heterozygous pathogenic NFIA variant in metopic craniosynostosis with preaxial polysyndactyly: A case report
Sipho Simon Nhongo, Eilidh Simpson, Moorthy Halsnad, Meharpal Sangra, Ahad Shafi, Jamie Campbell, Louise Thompson, David Moore

TL;DR
A new genetic variant in the NFIA gene is linked to a rare skull and hand deformity in a young boy.
Contribution
A novel heterozygous pathogenic NFIA variant is reported in a case of metopic craniosynostosis with preaxial polysyndactyly.
Findings
A novel NFIA variant was identified in a patient with metopic craniosynostosis and preaxial polysyndactyly.
The case highlights the rare association of NFIA-related disorder with craniosynostosis.
Surgical management and outcomes for the patient are detailed.
Abstract
Craniosynostosis is a congenital condition caused by the early fusion of one or more skull vault sutures during embryological development, resulting in an abnormal head shape. This condition has been linked to many gene variants. The authors report a case of a novel heterozygous pathogenic NFIA variant, in a young boy presenting with metopic craniosynostosis with preaxial polysyndactyly. Craniosynostosis is a rarely reported feature of Nuclear factor I/A (NFIA) related disorder. This report highlights the investigations that were performed to identify this variant and details the child’s surgical management and outcome.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
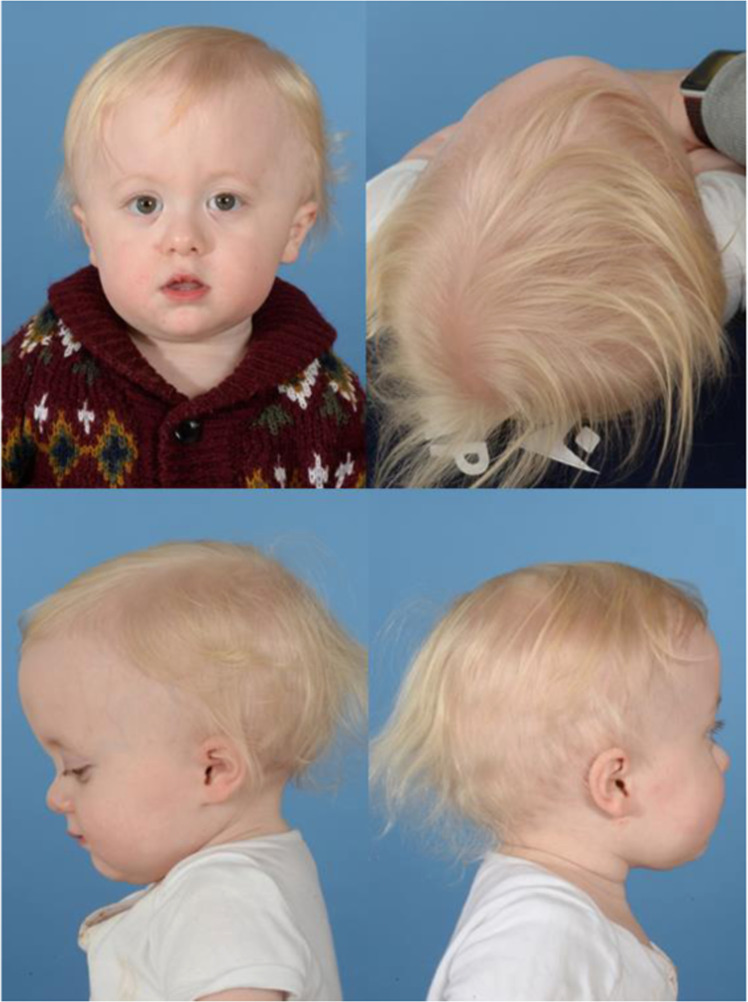 Figure 1
Figure 1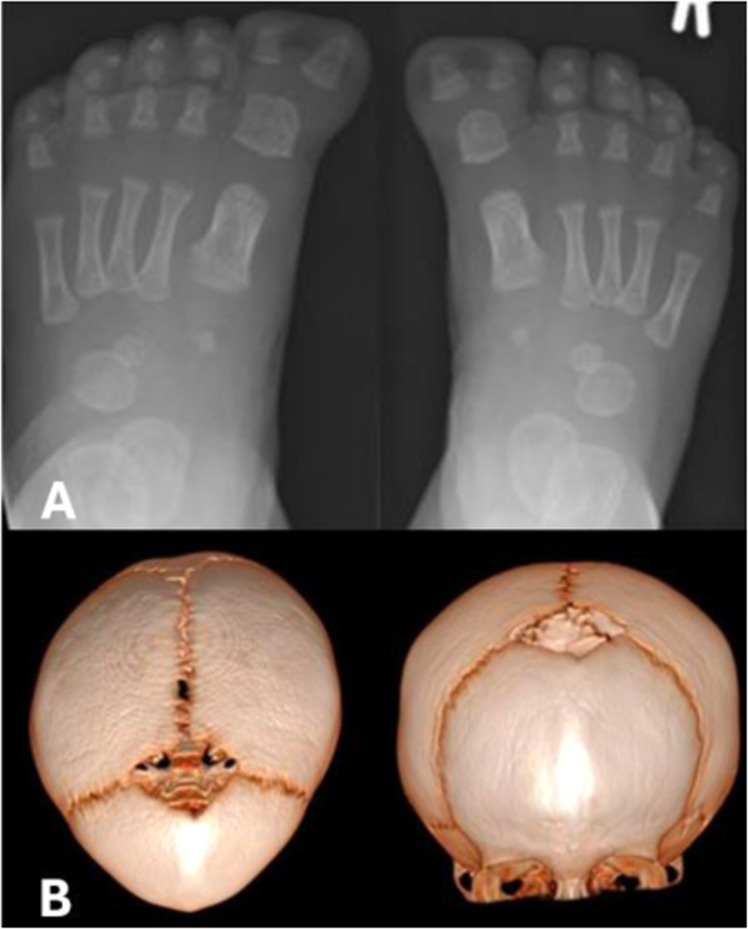 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCraniofacial Disorders and Treatments · Cleft Lip and Palate Research · Neurogenetic and Muscular Disorders Research
Introduction
Craniosynostosis is a congenital malformation caused by premature fusion of one or more skull vault sutures, resulting in abnormal head shape. Complications arise from compromised intracranial volume and may include raised intracranial pressure and visual impairment. Hearing loss or intellectual disability may be caused by the underlying gene defect or may occur secondarily. The global incidence is approximately 1–1.28 in 2,000 live births. Craniosynostosis can be further categorized into syndromic and non-syndromic types, with syndromic cases in the minority, accounting for 15–30 % of all presentations. Variants in at least 60 genes have been identified as recurrently causing craniosynostosis, and commonly implicated genes include EFNB1, ERF, FGFR2, FGFR3, SMAD6, TCF12 TWIST1.1
The Nuclear factor I/A (NFIA) gene (OMIM*600,727) is located on chromosome 1p31.3 and originates from the nuclear factor I (NFI) family of transcription factors. NFI proteins are crucial in central nervous system (CNS) development and functional faults may result in brain malformations, craniofacial and urinary tract anomalies.2^,^3 Haploinsufficiency of NFIA causes NFIA-related disorder (OMIM #613,735, officially named ‘brain malformations with or without urinary tract defects’ or (BRMUTD)) – a syndrome characterized by abnormalities such as macrocephaly, corpus callosum hypoplasia, hydrocephalus or ventriculomegaly, urinary tract defects, dysmorphic features, seizures and developmental delay. Less than thirty patients affected by this disorder have been identified and most arise from de novo variants.3
Craniosynostosis is a rarely reported feature of NFIA-related disorder, with only six documented cases–three of which were in a single family.4, 5, 6, 7
Polydactyly and polysyndactyly are rarely reported features of *NFIA-*related disorders. One article describes a 6-year-old boy who presented with preaxial polydactyly, bifid great toes, corpus callosum hypoplasia and macrocephaly with no craniosynostosis reported.2
There have been a few reports of genetic variants causing both polydactyly and craniosynostosis simultaneously, including FGFR, CRPT1 and CRPT2, leading to syndromes such as Apert syndrome and Carpenter syndrome. None of the current literature report a link between an NFIA variant resulting in both craniosynostosis and polydactyly.
Here, the authors present a case of an infant boy with metopic craniosynostosis, polysyndactyly and speech delay resulting from a de novo, pathogenic, heterozygous variant in the NFIA gene. This variant has not been reported in literature or within the gnomAD database.
Case report
The proband is a 9-week-old male who was referred to the Scottish National Craniofacial Service at the Royal Hospital for Children, Glasgow. Assessment was requested after parents and clinicians noted an abnormally large and misshapen head at birth.
The child was born at term to non-consanguineous parents, via C-section after prolonged induced labor. Birth weight was 3300 g (29th centile, Z −0.56). Clinical inspection at 8 months of age revealed a trigonocephalic appearance with a prominent metopic ridge, bitemporal pinching, hypotelorism and compensatory biparietal widening (See Figure 1). Macrocephaly was also noted, with an occipito-frontal circumference OFC >99th centile (49.5 cm at 8 months old(Z + 3.99) and 52 cm at 14 months (Z + 4.17)). The anterior fontanelle was open and normotensive.Figure 1. Preoperative clinical photographs at 21 months 19 days of age.Figure 1
Ophthalmological examination showed no evidence of raised intracranial pressure.
Limb inspection revealed bilateral great toe complex polysyndactyly (bifid great toes), with duplication of the distal phalanx of each great toe. While both thumbs appeared broad, x-rays showed no upper limb bony abnormalities.
Magnetic resonance imaging (MRI) at 2 months of age reported corpus callosum hypoplasia with dysgenesis. A frontal ridge suggestive of metopic synostosis was noted, however a computed tomography (CT) scan was suggested to better elucidate bony anatomy. Head CT scan was performed at 13 months and confirmed the findings.
Based on the findings of macrocephaly and polysyndactyly, genetic testing for Greig’s syndrome (cephalopolysyndactyly) was performed, however molecular analysis of GLI3 was normal. Additionally, on MLPA analysis of ROR2 and HOXD13, no deletion or duplication was detected Figure 2Figure 2(A) Left and right foot x-rays showing a duplicated distal phalanx of each great toe. (B) 3D reconstruction of computed tomography scan. Trigonocephalic appearance with triangular forehead, absent/fused metopic suture and metopic ridge.Figure 2
Trio-based whole exome sequencing was performed. Libraries were constructed using the Illumina DNA Prep with Enrichment kit and capture was performed using Human Core Exome and Human RefSeq Panel probes from Twist Bioscience. Libraries were sequenced on a NovaSeq 6000 instrument using an S2 flow cell (2 × 100 bp) (Illumina). Analysis was carried out applying the DDG2P gene panel (1936 Developmental Disorder Genotype-to-Phenotype genes).8 Confirmatory Sanger sequencing was performed. This identified the heterozygous, de novo, NFIA variant c.597dup p.(Ala200ArgfsTer2). The variant was assessed according to ACGS Best Practice Guidelines for Variant Classification in Rare Disease and was classified as pathogenic.9 The variant is predicted to result in a frameshift with the transcript likely to undergo nonsense-mediated decay (variant occurs in exon 3 of 11) and has not been reported within the Genome Aggregation Database (gnomAD, v4.1.0). NFIA nomenclature is based on NCBI reference sequence NM_001134673.4.
Considering this newly detected NFIA variant, a urinary tract ultrasound was performed, showing no abnormalities. Excision of duplicate great toe distal phalanges was performed at 12 months of age. Fronto-orbital advancement and remodelling (FOAR) to correct the trigonocephaly was performed at 21 months 20 days of age.
The child progressed well post-surgery, and at the time of writing, is in early years education receiving specialist input for delayed speech development. Supplementary material (figure 3) shows the child’s progress.
Discussion
Review of the existing literature consisted of a PubMed search using terms “nfia”, “variant”, “craniosynostosis”, “synostosis” and “nfia” in combination with Boolean logic. No restrictions on date of publication. The database search was complemented with manual review of the reference lists of relevant articles, which resulted in a few additional articles included in the study.
Table 1 details the six documented cases of craniosynostosis associated with an NFIA-related disorder.4, 5, 6, 7Table 1. Cases of Craniosynostosis associated with NFIA-related disorder.Table 1. AuthorsCaseNFIA variantPhenotypic traitsRao et al.,4Metopic synostosis in an 8-year-old femaleDe novo microdeletion of 120 Kilo base pair (Kb) in chromosome 1p31.3 region affecting exons four to nine; an discrete intragenic NFIA microdeletionMacrocephaly, hypoplasia of the corpus callosum, urinary tract abnormality (kinking of the right pelvi-ureteric junction) and developmental delayNyboe et al.,5Familial case of affecting four family members, with three out of four having craniosynostosis (two lambdoid, one sagittal).A 109Kb microdeletion of the gene affecting exons one and two was found in all family members.Hypoplasia of the corpus callosum, ventriculomegaly, developmental delay and renal tract abnormalitiesTonne et al.,6A 15-year-old boy who presented with metopic craniosynostosisTwo de novo NFIA variants found within exon 2 of the NFIA gene and resulted in induction of premature stop codonsThinning of the corpus callosum, macrocephaly and mild developmental delayBertini et al.,7A 2-year-old male toddler who presented with sagittal craniosynostosisA de novo 1.484 Kb NFIA gene deletionMacrocephaly, low-set ears, brachydactyly with bilateral proximally placed first fingers and short lower limbs, speech delay, corpus callosum hypoplasia and dysmorphic lateral ventricles
NFIA-related disorder is caused by a heterozygous inactivation or disruption of NFIA, without involvement of adjacent or surrounding genes. In addition to isolated NFIA disruption, deletion of NFIA can also occur as part of a multigenic microdeletion, such as 1p32-p31 deletion syndrome, of which there are less than ten cases reported. However, because NFIA is now widely viewed as the critical gene responsible for the 1p32-p31 deletion syndrome phenotypic traits, it is thought to be, itself, an NFIA-related disorder. Craniosynostosis has only on two occasions been described as a feature of 1p32-p31 deletion syndrome (with the NFIA gene encompassed by the multigenic deletion).10
The patient in the present study is unique not only in terms of the genetic variant, but also in terms of his phenotype. The patient exhibits classical features of a NFIA-related disorder (macrocephaly, corpus callosum hypoplasia and speech delay) whilst also presenting with preaxial polysyndactyly - only reported once before as a NFIA-related disorder and in that case, craniosynostosis was absent.9 Similarly, in NFIA-related disorder, metopic synostosis has only previously been reported twice.^12^ Overall, given the absence of seizures to date, and normal renal tracts on ultrasound, the newly detected NFIA variant c.597dup p.(Ala200fs) would appear to be associated with a milder phenotype of NFIA-related disorder. Genetic counselling will be required when this patient reaches young adulthood due to the autosomal dominant inheritance pattern of NFIA-related disorders.
Conclusion
This new NFIA variant is important for expanding the knowledge base of this rare condition, and the case strengthens the hypothesis that NFIA haploinsufficiency is a cause of craniosynostosis. This is supported by the decision of the NHS Genomic Medicine Service in England to ‘green-list’ the connection between syndromic craniosynostosis and the NFIA gene. Genetic testing for NFIA may be warranted in craniosynostosis presentations where the routine genetic tests are negative.
Declaration of competing interest
None of the authors involved in this case report have any competing interests to declare.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Tooze R.Calpena E.Weber A.Review of recurrently mutated genes in craniosynostosis supports expansion of diagnostic gene panels 1432023615 Accessed 13 May 2025. Available from:https://pubmed.ncbi.nlm.nih.gov/36980886/10.3390/genes 14030615 PMC 1004821236980886 · doi ↗ · pubmed ↗
- 2Paschell P.Laukaitis C.Significant phenotypic variability in a multigenerational family with an NFIA missense variant: case series and review of the literature Clin Case Rep 1212024264 Available fromhttps://pmc.ncbi.nlm.nih.gov/articles/PMC 10769898/10.1002/ccr 3.8307 PMC 1076989838188845 · doi ↗ · pubmed ↗
- 3Dini G.Verrotti A.Gorello P.NFIA haploinsufficiency: case series and literature review Front Pediatr 11202310.3389/fped.2023.1292654 PMC 1061684837915986 · doi ↗ · pubmed ↗
- 4Rao Anupam Ca SO’Donnell Bain N.Meldrum C.Shorter D.Goel H.An intragenic deletion of the NFIA gene in a patient with a hypoplastic corpus callosum, craniofacial abnormalities and urinary tract defects Eur J Med Genet 572–3201465702446288310.1016/j.ejmg.2013.12.011 · doi ↗ · pubmed ↗
- 5Nyboe D.Kreiborg S.Kirchhoff M.Hove H.B.Familial craniosynostosis associated with a microdeletion involving the NFIA gene Clin. Dysmorphol 24320151091122571455910.1097/MCD.0000000000000079 · doi ↗ · pubmed ↗
- 6Tonne E.Due-Tonnessen B.J.Mero I.L.Benefits of clinical criteria and high-throughput sequencing for diagnosing children with syndromic craniosynostosis 2020 Available from:https://www.nature.com/articles/s 41431-020-00788-410.1038/s 41431-020-00788-4PMC 818739133288889 · doi ↗ · pubmed ↗
- 7Bertini V.Cambi F.Orsini A.Phenotypic spectrum of NFIA haploinsufficiency: two additional cases and review of the literature Genes (Basel)1312202222493655351710.3390/genes 13122249 PMC 9777632 · doi ↗ · pubmed ↗
- 8Thormann A.Halachev M.Mc Laren W.Flexible and scalable diagnostic filtering of genomic variants using G 2P with Ensembl VEP Nat Commun 1012019237310.1038/s 41467-019-10016-331147538 PMC 6542828 · doi ↗ · pubmed ↗