Liver Transplantation for Isolated Langerhans Cell Histiocytosis in an Adult
Ruiyang Huang

TL;DR
A 56-year-old man with liver-only Langerhans cell histiocytosis underwent successful liver transplantation and remained stable for three years.
Contribution
This case report highlights liver transplantation as a potential treatment for isolated adult-onset LCH involving the liver.
Findings
The patient had unisystemic LCH limited to the liver, confirmed by pathological examination.
Liver transplantation led to stable liver function without recurrence or additional therapy for three years.
LCH was not detected in other systems, as confirmed by negative bone marrow and bone scan results.
Abstract
Langerhans cell histiocytosis (LCH) is a rare disease of proliferation of histiocytic disorder composed of histologically bland Langerhans cells mixed with reactive mononuclear and granulocytic cells, and often accompanied by eosinophils. These cells are characterized by expression of CD1a, S-100 and Langerin proteins. The clinical presentation ranges from indolent to aggressive, depending on the anatomic site involved which can be unifocal, multifocal, unisystemic, or multifocal and multisystemic disease. Cases involving LCH disease and treatment involving the liver are rare, especially in adult patients. Herein, we discuss a case of a 56-year-old male patient who presented with jaundice, acute abdominal pain, and a history of elevated liver function tests assumed to be caused by fatty liver disease. However, a computed tomography (CT) scan revealed a cholangiocarcinoma with associated…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
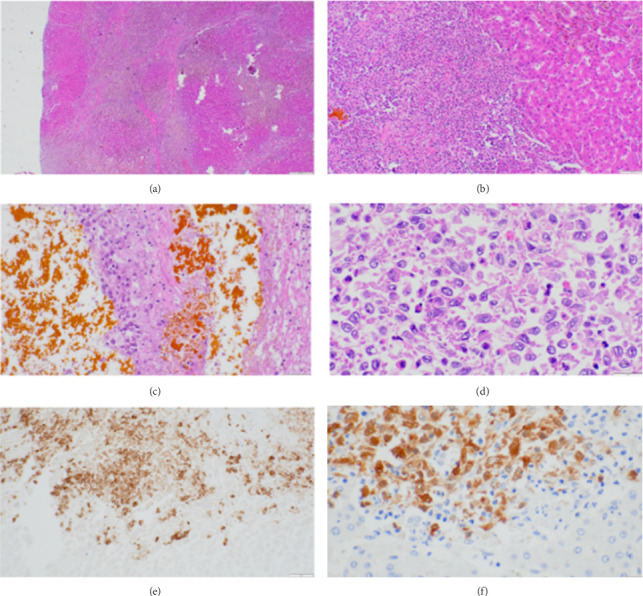 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHistiocytic Disorders and Treatments · Viral-associated cancers and disorders · Parvovirus B19 Infection Studies
1. Introduction
Langerhans cell histiocytosis (LCH) is the proliferation and accumulation of CD1a+ dendritic cells, known as Langerhans cells, and it typically manifests in one or more organ systems such as the skin, lung, bone, bone marrow, and liver [1, 2]. Involvement of LCH in the liver is considered “high risk,” as opposed to skin or bone involvement, and typically it manifests clinically with jaundice, fatigue, and pruritus in the early stages, and hepatomegaly, sclerosing cholangitis (SC), and liver failure in the late stages [1, 3]. Most cases occur in children, and it is more common in males than in females. Diagnosis of liver disease caused by LCH involvement is thought to be often unrecognized and unreported, especially amongst adults [1, 3, 4]. Initial diagnosis of LCH usually reports elevated liver function enzymes; however, the most definitive diagnosis can only be reached after liver biopsy. The presence of CD1a+ dendritic cells during pathological examination of liver biopsy samples is indicative of LCH involvement. Prior to the usage of orthotopic liver transplantation (LT) for LCH treatment, a majority (83%) of those suffering from LCH underwent chemotherapy; however, the prognosis of patients in the later stages with SC or cirrhosis was poor [3]. Upon review, only 10 other cases of liver disease caused by LCH involvement in adults have been found where orthotopic LT was pursued as a treatment option. Long-term follow-up of these patients will be indicative of the efficacy of LT as a treatment option for those patients in the late stages of liver disease caused by LCH.
2. Case Report
56-year-old male patient with history for hypertension, benign pituitary tumor, and “fatty liver” presented to our hepatology service in June 2018 for evaluation of elevated liver function tests (LFTs) and cirrhosis. He reportedly had a longstanding history of elevated LFTs as far back as 2012. The main abnormal LFTs are alkaline phosphatase elevation with progression up to 800 U/L, which had been assumed that “fatty liver” was the cause in the past. He presented to Emergency Department in January 2019 with acute abdominal pain and acute onset of jaundice. During his hospitalization abdominal magnetic resonance imaging (MRI) showed “mildly prominent intrahepatic ducts and abrupt termination of the common hepatic duct (CHD) with focal abnormal signaling concerning for cholangiocarcinoma/tumor.” A CT examination indicated “cholangiocarcinoma of the proximal to mid common bile duct (CBD) with associated biliary dilatation and metastatic peripancreatic/portal adenopathy, and cirrhosis.” Endoscopic retrograde cholangiopancreatography (ERCP) demonstrated “stricture in mid CBD with saccular dilation immediately above the stricture, and an additional stricture immediately above this saccular dilation, which appeared to be caused by extrinsic compression with lymph node.” The lymph node biopsy was negative for malignancy.
2.1. Diagnosis
Patient underwent exploratory laparotomy, cholecystectomy, and hepaticojejunostomy. Pathology examination showed the extrahepatic bile duct with involvement by LCH, marked chronic inflammation and fibrosis with mucosal ulceration. The wedge liver biopsy showed involvement by LCH, biliary fibrosis, cholestasis, and advanced fibrosis. Gallbladder demonstrated chronic cholecystitis.
Subsequent bone marrow biopsy was negative for LCH. Current recommendations published in 2022 suggest FDG-PET/CT for LCH patients to determine tumor staging [5], however this was not done in our patient. Instead, bone scan did not show any osseous involvement by LCH. Patient was diagnosed as LCH with isolated liver involvement and decompensated cirrhosis.
2.2. Treatment
Although Next Generation Sequencing (NGS) and targeted therapies are emerging treatments of histiocytic neoplasms, particularly for identifying and addressing MAPK pathway mutations [5], neither was considered or pursued in this case due to the patient's urgent clinical status and the immediate need for LT. LT was performed in July 2019. Pathology examination of the explanted liver showed LCH involvement with severe cholestasis and biliary type cirrhosis (Figure 1(a)). Portal areas showed mixed inflammation including lymphocyst, eosinophils, and Langerhans histiocytes (Figure 1(b)), bile ductular reaction, and severe SC with Langerhans histiocyte infiltrate with hepatolithiasis (Figure 1(c)). The Langerhans histiocytes demonstrated oval irregular folded nuclei, vesicular chromatin, inconspicuous nucleoli and ill defined, finely granular or vacuolated eosinophilic cytoplasm (Figure 1(d)), which were positive for S100 (Figure 1(e)) and CD1a (Figure 1(f)). No BRAF mutation was detected by molecular tests in this case.
2.3. Outcomes
The patient has been undergoing surveillance without any therapy for LCH. His liver function has been stable for 3.5 years with no notable adverse and unanticipated events relating to his liver transplant.
3. Discussion
Although liver involvements in LCH cases are rare, reported as 10.1%–18% in pediatric patients with LCH, and cases of hepatic involvement in adults leading to SC are rarely reported and reviewed [2]. Disease prognosis is significantly worse with liver involvement; a fatality rate of 30%–50% is observed versus less than 10% when the liver is not involved [1, 3].
Diagnosis of this disease is also difficult without liver biopsy. LCH shares common clinical and pathological symptoms with many liver diseases such as jaundice, cholangitis, cirrhosis, pruritus, fatigue, and elevated LFTs. In our case, our patient had presented a year earlier with elevated liver function enzymes and it was assumed that these were only the result of his fatty liver disease. Only after follow-up and the presence of later stage symptoms such as jaundice and SC was a definitive diagnosis of LCH reached after immunohistochemical analysis proved the presence of CD1a+ Langerhans cells in the patient's liver biopsy.
As for treatment of unisystemic LCH in the liver or spleen, systemic therapies including cladribine, cytarabine, or vinblastine and prednisone are recommended while smoking cessation is the first line recommendation for unisystemic pulmonary LCH [5]. However, LT has shown success specifically in treating LCH involving end-stage liver disease involving cirrhosis [6–8].
Ziogas et al. [4] reviewed LT cases from 1987 to 2018 in a US population-based analysis containing a total of 60 patients with secondary SC due to LCH undergoing LT. The median age at LT was 3.0 years of age. LT recipients' survival outcomes were acceptable and the disease recurrence rate to the liver was low. The conclusion was that LT should be considered in patients with liver failure secondary to LCH even in the setting of active multisystem disease, if recipients are expected to have good survival and options for salvage therapy in the setting of disease reactivation. Of note, only 5 patients were adults, and the clinical presentation of these cases was not documented in this review. To the best of our knowledge, there were only 10 adult LCH patients treated with LT from 1990 to present. The clinical features, prognosis, and treatment are summarized in Table 1.
As seen in Table 1, even among this handful of cases lie differences in patient symptoms, treatment methods, and treatment outcomes. For example, the current standard of care for multisystemic LCH includes systemic chemotherapy (vinblastine, prednisone, and mercaptopurine) [15, 16], whereas the standard for unifocal LCH has been surgical resection [17]. Analysis of long-term outcomes of LCH showed a 5-year overall survival (OS) rate of 88% with these first-line treatments; however, when a “high risk organ,” such as the liver or spleen, was involved, their OS rate worsened (Hazard Ratio, 10.8; 95% CI, 3.3–35.5; p < 0.001), and patients were 10.8 times more likely to not have survived at 5 years [17]. Interestingly, in two of the cases reviewed, chemotherapeutics either worsened the severity of the patient's liver injury [9] or did not have any effect on the patient's liver damage [8]. Whereas in another case, recurrence of LCH was successfully controlled using chemotherapy [11]. Fortunately, a similarity found among these cases is the scarcity of LCH recurrence at follow-up. Among these cases, only three have recorded follow-ups with a longer post-operation duration than our patient. Our current case adds to the small list of LCH patients who have undergone LT in adults, and long-term follow-up of these patients will be the key to understanding the extended efficacy of transplantation as a treatment for LCH-related liver disease.
3.1. Learning Points
Although rare, LCH involvement in the liver is a serious concern for pediatric and adult patients with LCH alike, as its involvement in the liver greatly increases the fatality rate of LCH. Diagnosis of LCH is complicated by similarities in clinical symptoms with many other liver diseases such as fatty liver disease or primary SC. The main differentiator between LCH and other liver diseases that also result in end-stage liver disease is the presence of CD1a+ dendritic cells, especially proximal to the bile ducts. Orthotopic LT has emerged as a possible treatment option, especially for patients in decompensated cirrhosis secondary to liver involvement. There are very few other patients that have been monitored for a longer duration than our patient after LT for LCH liver disease, and the relative absence of recurrence of liver disease in these patients is a promising sign of the efficacy of LT as a treatment for LCH related liver disease.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Fu Z. Li H. Arslan M. E. Ells P. F. Lee H. Hepatic Langerhans Cell Histiocytosis: A Review World Journal of Clinical Oncology 202112533534110.5306/wjco.v 12.i 5.33534131565 PMC 8173326 · doi ↗ · pubmed ↗
- 2Hatemi I. Baysal B. Senturk H. Behzatoglu K. Bozkurt E. R. Ozbay G. Adult Langerhans Cell Histiocytosis and Sclerosing Cholangitis: A Case Report and Review of the Literature Hepatology International 20104365365810.1007/s 12072-010-9205-32-s 2.0-7795810719321063491 PMC 2940001 · doi ↗ · pubmed ↗
- 3Abdallah M. Généreau T. Donadieu J. Langerhans’ Cell Histiocytosis of the Liver in Adults Clinics and Research in Hepatology and Gastroenterology 2011356-747548110.1016/j.clinre.2011.03.0122-s 2.0-8005231146521550330 · doi ↗ · pubmed ↗
- 4Ziogas I. A. Kakos C. D. Wu W. K. Liver Transplantation for Langerhans Cell Histiocytosis: A US Population‐Based Analysis and Systematic Review of the Literature Liver Transplantation 20212781181119010.1002/lt.2599533484600 · doi ↗ · pubmed ↗
- 5Goyal G. Tazi A. Go R. S. International Expert Consensus Recommendations for the Diagnosis and Treatment of Langerhans Cell Histiocytosis in Adults Blood 2022139172601262110.1182/blood.202101434335271698 PMC 11022927 · doi ↗ · pubmed ↗
- 6Griffiths W. Davies S. Gibbs P. Thillainayagam A. Alexander G. Liver Transplantation in an Adult With Sclerosing Cholangitis Due to Langerhans Cell Histiocytosis Journal of Hepatology 200644482983110.1016/j.jhep.2005.12.0242-s 2.0-3364491643916483684 · doi ↗ · pubmed ↗
- 7Lee R. J. Leung C. Lim E. J. Liver Transplantation in an Adult With Sclerosing Cholangitis Due to Multisystem Langerhans Cell Histiocytosis American Journal of Transplantation 20111181755175610.1111/j.1600-6143.2011.03661.x 2-s 2.0-7996108116821797977 · doi ↗ · pubmed ↗
- 8Scanzi J. Goutte M. Teilhet C. Abergel A. When Should We Consider Transplantation in Adult Patients With Sclerosing Cholangitis Due to Multisystem Langerhans’ Cell Histiocytosis? Digestive and Liver Disease 201547217617710.1016/j.dld.2014.09.0212-s 2.0-8492128557125455153 · doi ↗ · pubmed ↗