Extramedullary Involvement in T/Myeloid Mixed Phenotype Acute Leukemia With BCR::ABL1 Fusion in a Hispanic Female Patient: A Case Report
Camille L Santiago-Negron, José M Cordero Hernández, William D Marrero-León, María D Rivera Rolón

TL;DR
A rare case of T/myeloid mixed phenotype acute leukemia with BCR::ABL1 fusion and extramedullary involvement is reported in a 61-year-old Hispanic woman.
Contribution
This case report highlights the diagnostic challenges of a rare leukemia subtype with extramedullary presentation and BCR::ABL1 fusion.
Findings
A 61-year-old Hispanic female presented with generalized lymphadenopathy and extramedullary disease.
The case exhibited T/myeloid MPAL with BCR::ABL1 fusion detected via FISH analysis.
Integrated morphology, immunophenotyping, and molecular testing were critical for diagnosis.
Abstract
Mixed phenotype acute leukemia (MPAL) is a rare subtype of acute leukemia characterized by the expression of markers from more than one lineage. The T/myeloid subtype, especially with extramedullary involvement and BCR::ABL1 fusion, is exceptionally rare and diagnostically challenging. We report a case of a 61-year-old Hispanic female patient presenting with generalized lymphadenopathy. Excisional biopsy of a left occipital lymph node showed diffuse effacement by a monomorphic population of blasts. Immunohistochemistry revealed co-expression of myeloid (partial MPO, lysozyme) and T-lineage markers (CD3, CD4, CD5, CD7). Bone marrow biopsy confirmed MPAL with extensive infiltration by CD3+, CD5+, and CD2+ cells; approximately 20-30% co-expressed CD34, CD117, and TdT. Flow cytometry supported a diagnosis of T/Myeloid MPAL. Fluorescence in situ hybridization (FISH) analysis identified…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
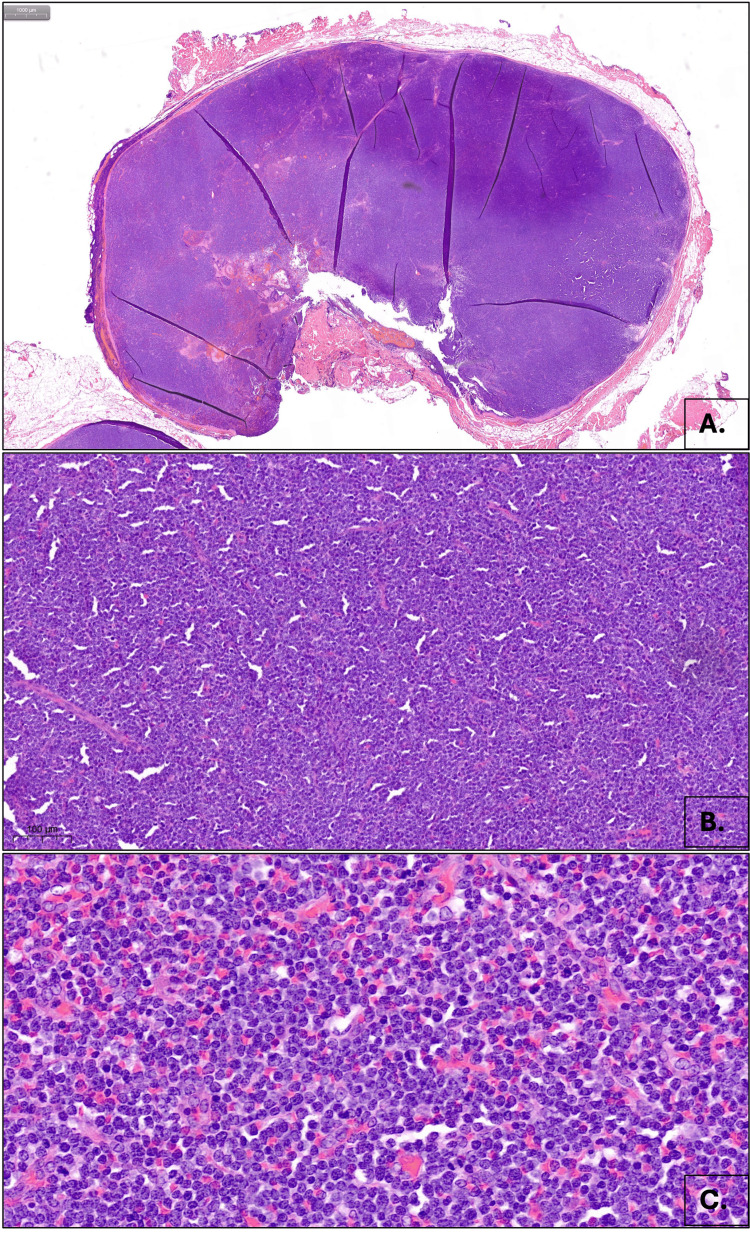 Figure 1
Figure 1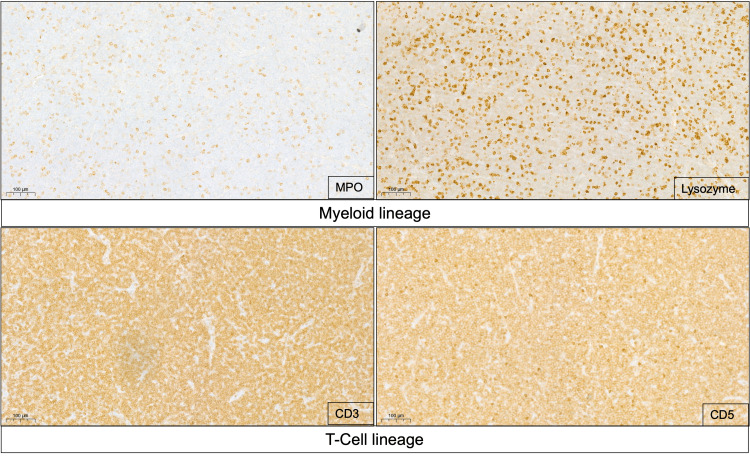 Figure 2
Figure 2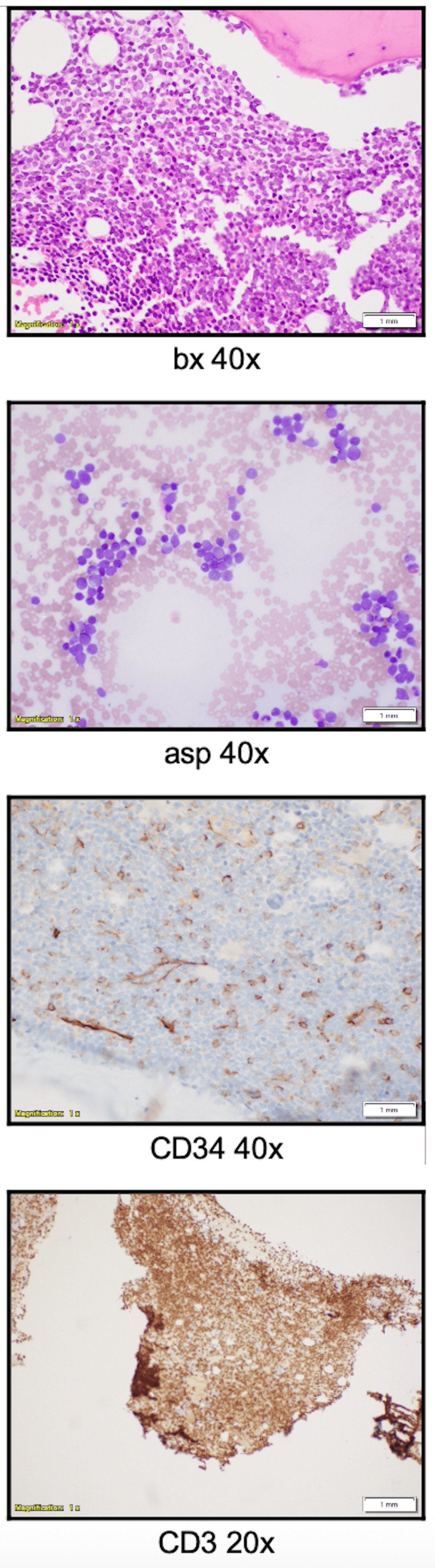 Figure 3
Figure 3| Marker | Result |
| CD3 | Positive (cytoplasmic) |
| CD4 | Positive |
| CD5 | Positive |
| CD7 | Positive |
| CD34 | Positive |
| CD117 (CKIT) | Positive |
| MPO | Positive (subset) |
| Lysozyme | Positive (subset) |
| CMYC | Overexpressed |
| KI-67 | ~100% |
| BCL2 | Positive (diffuse) |
| CD10 | Negative |
| CD15 | Positive (rare) |
| CD20 | Positive (residual B cells) |
| CD21 | Positive (residual FDC meshwork) |
| CD30 | Negative |
| CD68 | Positive |
| CD138 | Positive (rare) |
| Cyclin D1 | Negative |
| MUM1 | Positive |
| PAX5 | Positive (residual) |
| CD8 | Positive (rare) |
| EBER (EBV ISH) | Negative |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAcute Myeloid Leukemia Research · Chronic Myeloid Leukemia Treatments · Acute Lymphoblastic Leukemia research
Introduction
Mixed phenotype acute leukemia (MPAL) is a rare and biologically heterogeneous group of leukemias characterized by blasts co-expressing markers of different lineages, including myeloid, B-lymphoid, and T-lymphoid antigens. It accounts for approximately 2%-5% of all acute leukemias and poses significant diagnostic and therapeutic challenges [1,2]. The World Health Organization (WHO) classifies MPAL based on immunophenotypic and molecular features, including subtypes such as B/Myeloid and T/Myeloid [3].
MPAL with a T/Myeloid phenotype is particularly uncommon, and extramedullary involvement, especially with nodal disease, is even rarer. MPAL with BCR::ABL1 fusion is associated with a poor prognosis and requires targeted therapy with tyrosine kinase inhibitors (TKIs) alongside intensive chemotherapy [4,5]. We report a case of MPAL (T/Myeloid) with BCR::ABL1 fusion with generalized lymphadenopathy as the primary presentation, emphasizing the histopathological, immunophenotypic, and molecular features crucial for diagnosis.
Case presentation
Clinical presentation
A 61-year-old female patient presented with progressive, painless generalized lymphadenopathy. Physical examination revealed palpable lymph nodes in the cervical, axillary, and inguinal regions. She denied constitutional symptoms such as fever, night sweats, or weight loss. Laboratory evaluation showed significant leukocytosis (white blood cell count (WBC) 75.2K/µL) with circulating blasts (45%) and lymphocytosis (42%). Normocytic-normochromic Anemia (hemoglobin (Hb) 10.8 g/dL) and (platelets 211K/µL) were also noted.
Histopathology
Histopathologic examination demonstrated complete effacement of the normal lymph node architecture by a diffuse proliferation of medium to large monomorphic cells arranged in sheets. The neoplastic cells exhibited dispersed chromatin, inconspicuous nucleoli, and a high mitotic index (six mitoses per high-power field). Scattered residual secondary germinal centers were noted at the cortex (Figure 1A-C).
Extramedullary involvement in MPAL.A. Diffuse effacement of lymph node architecture, at 20x magnification, B. Sheets of medium to large monomorphic cells, at 200x magnification. C. Cytologic features demonstrating dispersed chromatin, inconspicuous nucleoli, and high mitotic index, at 600x magnification.MPAL: mixed phenotype acute leukemia.
Immunohistochemistry of the excised lymph node revealed that the neoplastic cells expressed CD34, c-KIT (CD117), CD3, CD4, CD5, CD7, myeloperoxidase (MPO, partial), and lysozyme. TdT was only expressed on rare cells, supporting a diagnosis of extramedullary involvement by T/Myeloid MPAL (Figure 2 and Table 1).
MPAL Myeloid/T-cell neoplastic lymph node infiltrate.MPO and lysozyme-positive staining markers of myeloid lineage. CD3 and CD5 positive staining markers of T cell lineage.MPAL: mixed phenotype acute leukemia.
A bone marrow biopsy demonstrated hypercellularity (30%) with 77% blasts, an immature infiltrate, and residual trilineage hematopoiesis. Immunostains confirmed extensive marrow infiltration by CD3+, CD5+, and CD2+ cells (90% of all cells). Additionally, approximately half of the abnormal cells (20-30%) expressed CD34, CD117, and TdT, further supporting a T/Myeloid MPAL phenotype (Figure 3).
Bone marrow biopsy showing blast population expressing CD34 and cytoplasmic CD3.
Flow cytometry of the bone marrow aspirate revealed a blast population (22.4%) expressing CD34, cytoplasmic CD3, partial CD117, partial MPO, CD7, and CD56, while being negative for TdT.
Cytogenetic and molecular studies identified a BCR-ABL1 [t(9;22)] gene rearrangement in 10% of cells by fluorescence in situ hybridization (FISH) analysis, with negative results for promyelocytic leukemia/retinoic acid receptor alpha (PML-RARA), core binding factor subunit beta (CBFB), lysine methyltransferase 2A (KMT2A) (MLL), and RUNX1T1/RUNX1. Cytogenetic analysis was inconclusive due to insufficient metaphases, and next-generation sequencing (NGS) (Tempus, Chicago, IL) did not identify significant pathogenic mutations in addition to the actionable mutation in BCR-ABL1.
Treatment
MPAL is particularly difficult to treat, in part because it is unclear whether it should be approached as acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), or a combination of both [5]. This uncertainty complicates optimal clinical care. Based on the lack of clear treatment regimens, we decided to treat our patient with a combined regimen using fludarabine, cytarabine, idarubicin, granulocyte colony-stimulating factor (GCF), and venetoclax (FLAG-IDA + VEN). The FLAG-IDA + VEN regimen is an active treatment for AML, capable of producing high remission rates and facilitating transition to allogeneic hematopoietic stem cell transplantation (alloHSCT) when appropriate for most patients. A recent publication by the MD Anderson group revealed an overall response rate above 90% with the FLAG-IDA + VEN regimen [6,7]. Since our patient also presented with BCR-ABL positivity, we decided to add targeted therapy with dasatinib to prepare for an allogeneic hematopoietic stem cell transplant.
Discussion
MPAL is a rare and diagnostically challenging entity. The T/Myeloid subtype represents a small fraction of cases and is often associated with an aggressive clinical course [1,4]. Extramedullary involvement in MPAL is rare, with nodal disease being an unusual presentation. This case underscores the importance of considering MPAL in patients presenting with generalized lymphadenopathy and circulating blasts.
An important differential diagnosis in this case is early T-cell precursor acute lymphoblastic leukemia (ETP-ALL), which shares significant immunophenotypic overlap with MPAL of T/Myeloid lineage. Both entities may express cytoplasmic CD3 and aberrant myeloid or stem cell markers, such as CD34, CD117, CD13, and CD33. However, ETP-ALL is defined by the absence of CD1a and CD8, weak or absent expression of CD5, and the presence of two or more myeloid or stem cell markers (e.g., CD13, CD33, CD34, CD117, HLA-DR, CD11b), without fulfilling the full criteria for myeloid lineage assignment [2]. In contrast, the diagnosis of MPAL requires definitive evidence of both T-lineage (cytoplasmic CD3) and myeloid-lineage (cytoplasmic myeloperoxidase or clear monocytic differentiation), which was evident in our case. Additionally, the presence of the* BCR::ABL1* fusion supports the classification of this leukemia as MPAL (T/myeloid) with a defined genetic abnormality, in accordance with the World Health Organization (WHO), fifth edition, and the International Consensus Classification (ICC) [1,8].
MPAL with* BCR::ABL1* fusion is particularly significant due to its prognostic and therapeutic implications. The optimal treatment strategy remains unclear, as MPAL with BCR::ABL1 fusion is associated with poor responses to standard regimens and a high relapse rate [5,6]. BCR::ABL1 rearrangements are among the most common genetic abnormalities in MPAL, present in approximately 14%-25% of the cases [1,9]. Their presence has significant therapeutic implications, as studies have shown improved outcomes when tyrosine kinase inhibitors (TKIs), such as imatinib or dasatinib, are incorporated into treatment regimens [9]. The management of MPAL remains particularly challenging due to its rarity, biological heterogeneity, and the lack of consensus on whether to treat it with AML, ALL, or hybrid protocols. Reported complete remission rates vary widely, ranging from 30% to 85%, and overall prognosis tends to be poorer than for other acute leukemias [9,10].
Several retrospective studies suggest that ALL-like regimens may offer improved outcomes, especially when combined with TKIs in BCR::ABL1-positive cases [9,10]. More recently, personalized approaches integrating cytogenetic, molecular, and immunophenotypic data, with allogeneic hematopoietic stem cell transplantation (allo-HSCT) in first remission when feasible, are increasingly being adopted. In the case we report, this evidence-based rationale informed the therapeutic approach: the patient was treated with dasatinib, a* BCR::ABL1*-targeted TKI, as a preparatory strategy for allo-HSCT (as detailed in the Treatment section), reflecting current practice patterns for this high-risk leukemia subtype.
This case highlights the diagnostic role of immunophenotyping and molecular studies in distinguishing MPAL from other acute leukemias and the importance of an integrated diagnosis between the medullary and the extramedullary findings. The presence of CD34, cytoplasmic CD3, and partial myeloid markers (CD117, MPO) was crucial in classifying this leukemia as T/Myeloid MPAL. Furthermore, the presence of TdT in rare cells, a marker typically seen in lymphoblastic leukemias, reinforces the unique immunophenotypic profile of this case.
Early recognition of extramedullary MPAL, particularly in the setting of lymphadenopathy, is essential for prompt diagnosis and treatment initiation. Further studies are needed to determine the most effective therapeutic strategies for MPAL with BCR::ABL1 fusion, especially in cases with nodal involvement.
Conclusions
This case emphasizes the importance of a multimodal approach to diagnosing MPAL, particularly in patients with atypical presentations such as generalized lymphadenopathy. The combination of histopathology, immunophenotyping, and molecular studies was essential in establishing the diagnosis. Given the aggressive nature of MPAL with BCR::ABL1 fusion, early diagnosis and targeted therapy with TKIs are critical. Looking ahead, the integration of novel tools such as digital pathology platforms and machine learning algorithms may enhance the early detection of genomic abnormalities and assist in predicting treatment responses, potentially reducing turnaround times and accelerating the initiation of tailored therapies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data Blood Arber DA Orazi A Hasserjian RP 1200122814020223576789710.1182/blood.2022015850 PMC 9479031 · doi ↗ · pubmed ↗
- 2The genetic basis and cell of origin of mixed phenotype acute leukaemia Nature Alexander TB Gu Z Iacobucci I 37337956220183020939210.1038/s 41586-018-0436-0PMC 6195459 · doi ↗ · pubmed ↗
- 3Mixed-phenotype acute leukemia: historical overview and a new definition Leukemia Weinberg OK Arber DA 184418512420102084456610.1038/leu.2010.202 · doi ↗ · pubmed ↗
- 4Genomic landscape of mixed phenotype acute leukemia associated with immunophenotypic lineage predominance: impact on diagnosis and treatment Eur J Haematol Zheng R Gagan JR Botten GA 1041105111420254009840910.1111/ejh.14414 · doi ↗ · pubmed ↗
- 5How to think about acute leukemia of ambiguous lineage Hematology Am Soc Hematol Educ Program Weinberg OK 287292202420243964401410.1182/hematology.2024000554 PMC 11665627 · doi ↗ · pubmed ↗
- 6Philadelphia chromosome-positive mixed phenotype acute leukemia in the imatinib era Eur J Haematol Shimizu H Yokohama A Hatsumi N Takada S Handa H Sakura T Nojima Y 2973019320142475030710.1111/ejh.12343 · doi ↗ · pubmed ↗
- 7Long term results of venetoclax combined with FLAG-IDA induction and consolidation for newly diagnosed and relapsed or refractory acute myeloid leukemia Leukemia Di Nardo CD Jen WY Takahashi K 8548633920254000084210.1038/s 41375-025-02531-8PMC 11978487 · doi ↗ · pubmed ↗
- 8The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms Leukemia Khoury JD Solary E Abla O 170317193620223573283110.1038/s 41375-022-01613-1PMC 9252913 · doi ↗ · pubmed ↗