Identification and characterization of nine PAT1 genes subfamily in Medicago edgeworthii
Gaoping Tang, Tingting Ni, García-Caparrós Pedro, Li-Hua Meng, Xudong Sun

TL;DR
This study identifies and characterizes nine PAT1 subfamily genes in Medicago edgeworthii, revealing their role in plant development and stress response.
Contribution
The first comprehensive characterization of PAT1 subfamily genes in Medicago edgeworthii.
Findings
Nine PAT1 subfamily genes were identified in Medicago edgeworthii with a conserved GRAS domain.
PAT1 subfamily genes are expressed in roots, stems, and leaves, suggesting broad developmental roles.
PAT1 proteins activate the MeDOF3.4 gene, potentially promoting cell proliferation and graft fusion.
Abstract
GRAS proteins represent a unique class of transcription factors that are exclusive to plants. Among the various subfamilies within the GRAS family, the phytochrome A signal transduction 1 (PAT1) subfamily is particularly prominent, given its multifaceted regulatory functions in phytochrome signaling pathways and stress response mechanisms, as well as its involvement in plant developmental processes. Despite the recognized importance of GRAS proteins, there are no studies to date that have characterized the GRAS gene family in Medicago edgeworthii. In this study, we performed a comprehensive genome-wide analysis of GRAS genes and identified nine genes belonging to the PAT1 subfamily in M. edgeworthii. Multiple sequence alignment of these proteins revealed the presence of a conserved C-terminal GRAS domain, alongside a highly variable N-terminal region. Additionally, we observed that…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
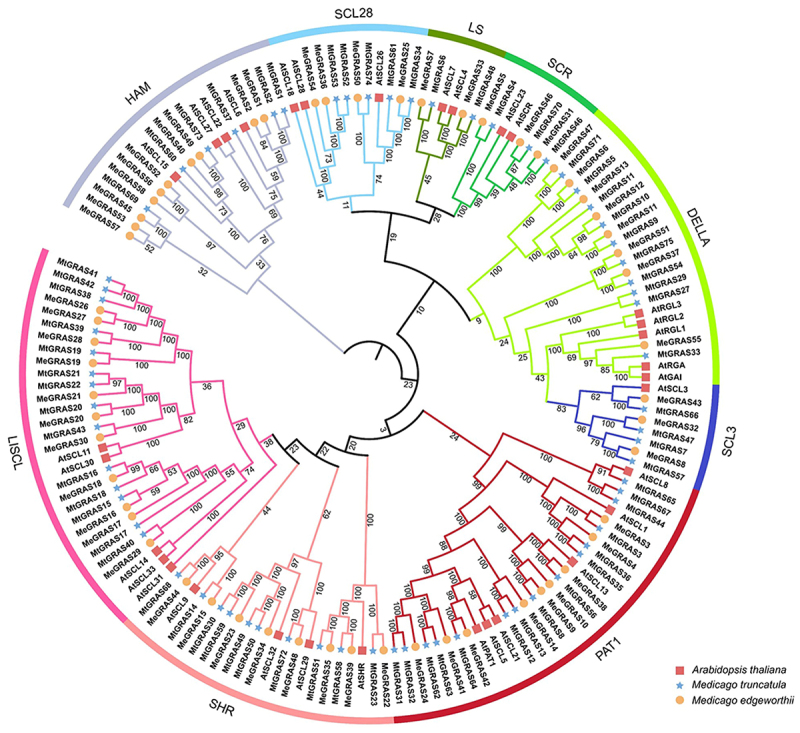 Figure 1
Figure 1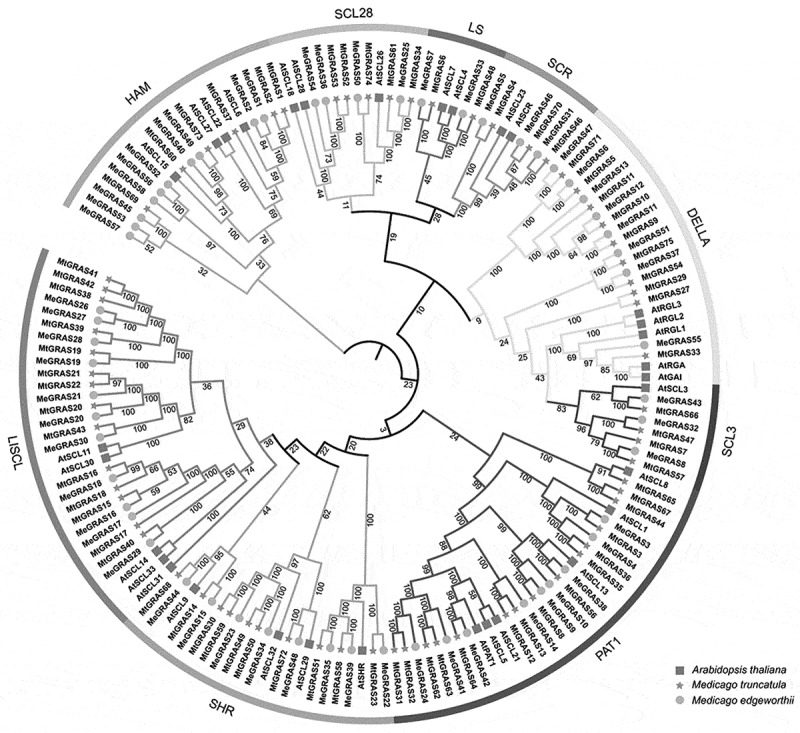 Figure 2
Figure 2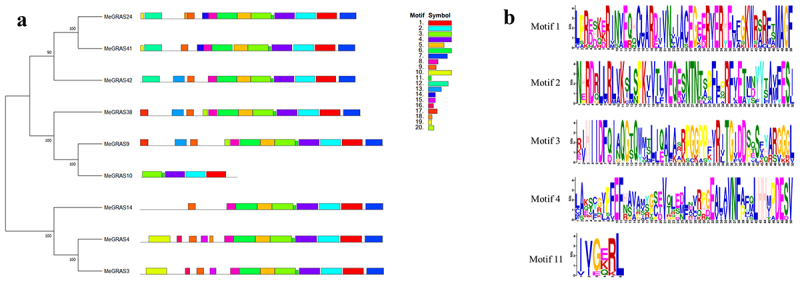 Figure 3
Figure 3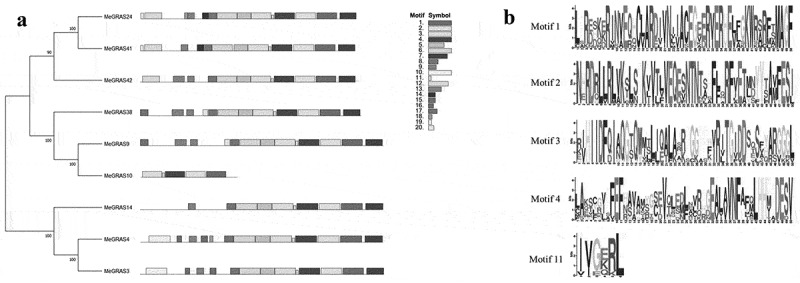 Figure 4
Figure 4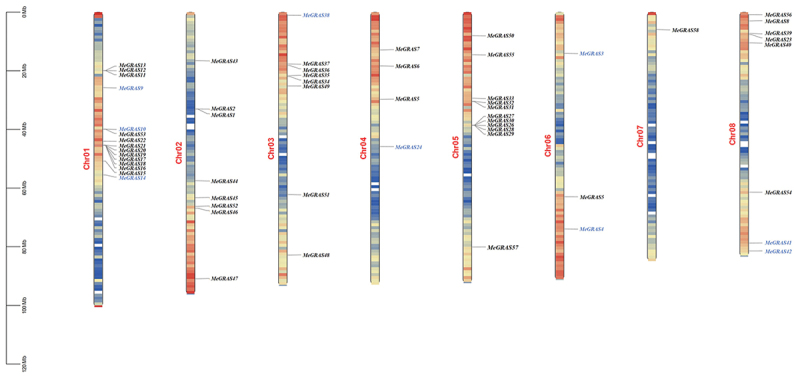 Figure 5
Figure 5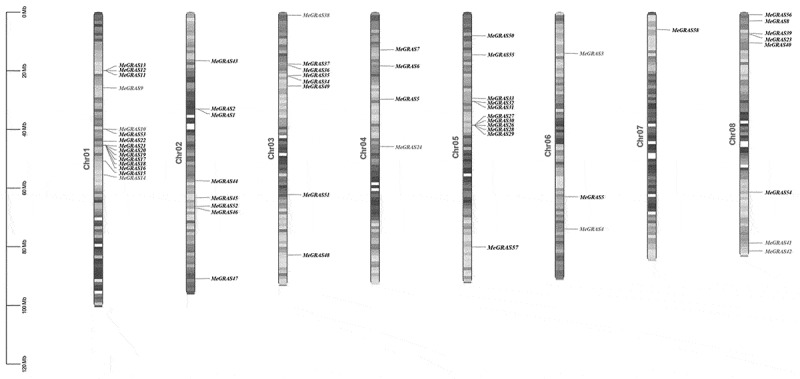 Figure 6
Figure 6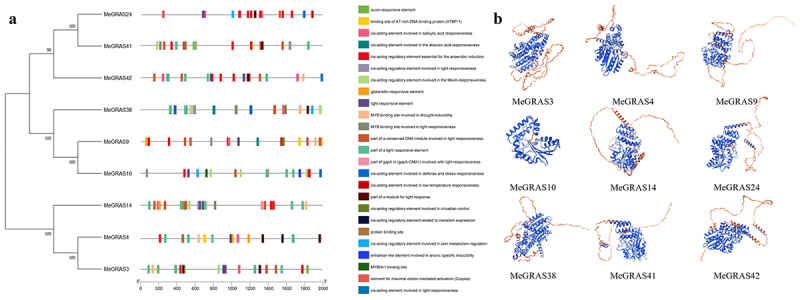 Figure 7
Figure 7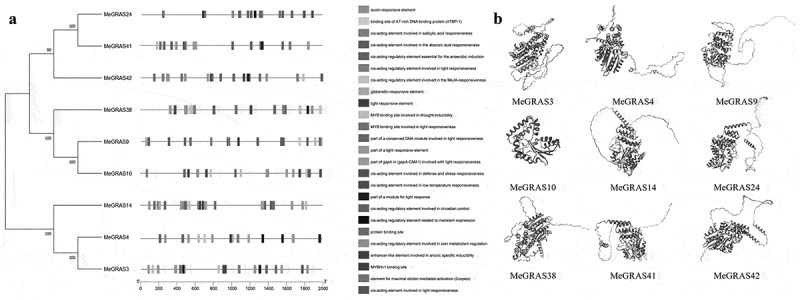 Figure 8
Figure 8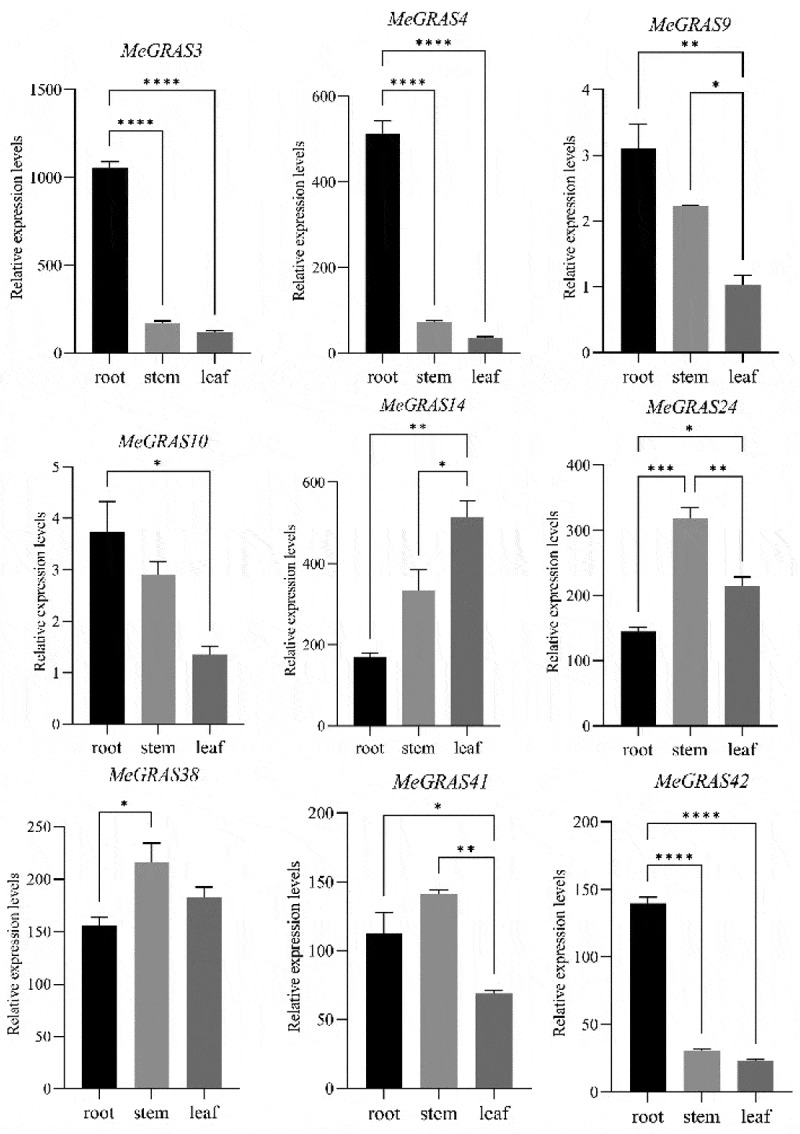 Figure 9
Figure 9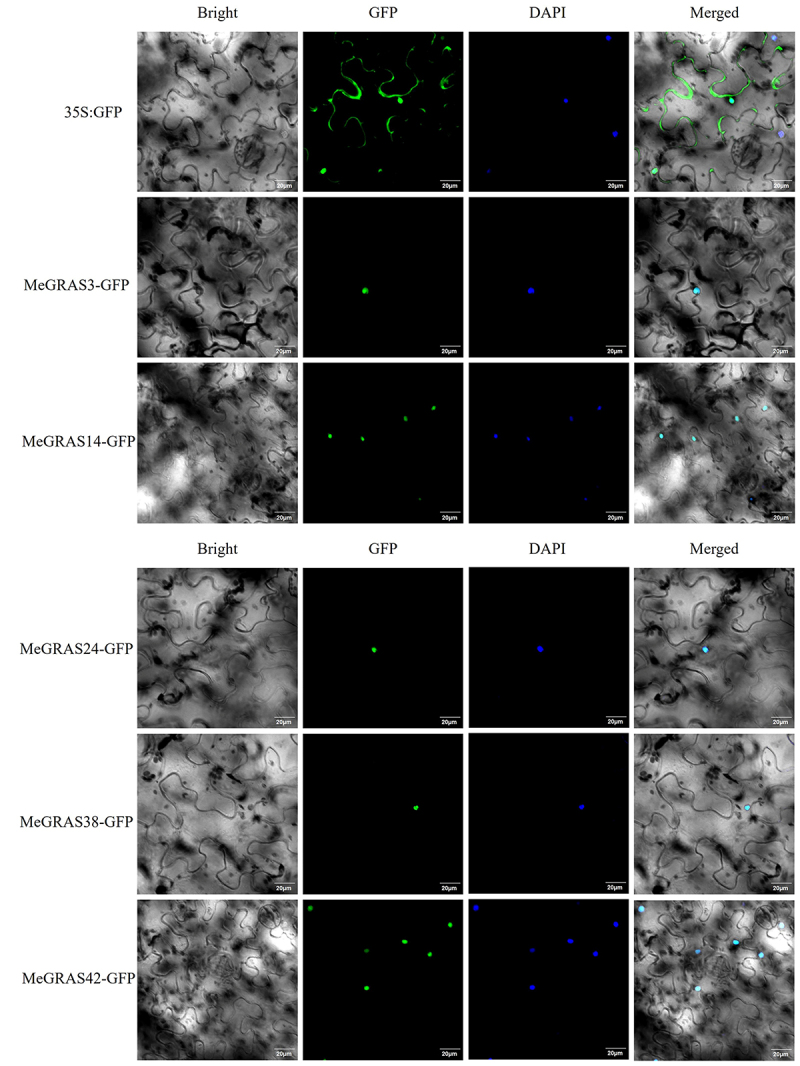 Figure 10
Figure 10 Figure 11
Figure 11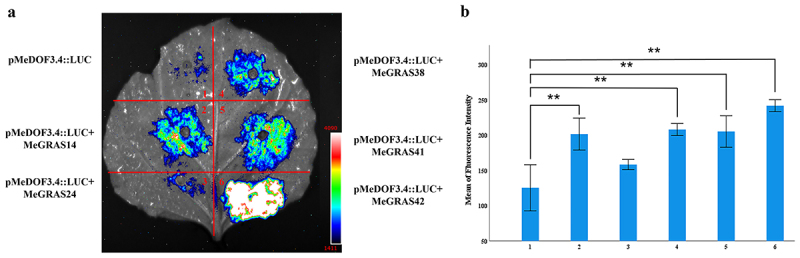 Figure 12
Figure 12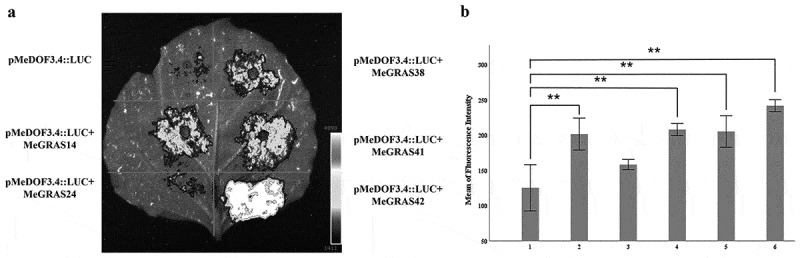 Figure 13
Figure 13- —Basic Research Project of Yunnan
- —National Natural Science Foundation of China10.13039/501100001809
- —National Natural Science Foundation of China10.13039/501100001809
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Reproductive Biology · Plant-Microbe Interactions and Immunity · Plant nutrient uptake and metabolism
Introduction
The GRAS protein family comprises a group of plant-specific transcription factors, named after its three originally identified family members Gibberellin acid insensitive (GAI), Repressor of GA1 (RGA) and SCARECROW (SCR).^1–4^ Typical GRAS proteins usually comprise 400 ~ 770 amino acid residues and are characterized by a highly conserved C-terminal region and a variable N-terminal region.^5^ The highly conserved C-terminus of GRAS proteins can be divided into five different motifs based on their amino acid compositions:, LHR I, VHIID, LHR II, PFYRE, RVER, and SAW.^6^ Among these, the VHIID conserved sequence is the core structure of the GRAS proteins and is present across all members of the family.^7^ According to different protein structural characteristics, the GRAS family is further categorized into eight subfamilies: DELLA, LAS, SCR, SHR, PAT1, HAM, LISCL, and SCL3.^8–10^
The PAT1 subfamily is a subgroup within the GRAS transcription factor families.^2,11^ There are six PAT1 subfamily members identified in Arabidopsis, including AtPAT1, AtSCL1, AtSCL5, AtSCL8, AtSCL13, and AtSCL21.^12^ Among these, AtPAT1, AtSCL5, AtSCL13 and AtSCL21 are highly homologous and play important roles in regulating the phytochrome signal transduction,^13–15^ responding to abiotic stress^16–18^ and maintaining meristematic tissues.^19,20^ Specifically, AtPAT1, AtSCL5, and AtSCL21 are positive regulators in the phytochrome A signaling pathway,^11,14^ and they share a common EAISRRDL protein motif at their N terminus, a feature which is absent in other members of the family. Another member of the PAT1 subfamily, SCL13, acts as a positive regulator in the red light signaling pathway downstream of phytochrome B,^15^ playing an essential role in the elongation of hypocotyl. Moreover, members of the PAT1 subfamily have been implicated in wound healing during grafting. PAT1 has been identified as a key regulator of graft healing in both Picea abies and Arabidopsis thaliana. Notably, Arabidopsis thaliana mutants lacking the PAT1 gene could not be successfully grafted,^20^ whereasAtPAT1 over-expressing leads to increased callus formation at the site of petiole injury.^20^ Additionally, the PAT1-ERF115 protein complex is pivotal for maintaining the function of meristematic tissues by promoting the regeneration of damaged stem cells.^21^
The DNA-binding with one finger (Dof) family represents a unique class of transcription factors exclusive to plants, belonging to the zinc finger protein subfamily.^22^ Among them, the DOF3.4 transcription factor (also known as OBP1) in Arabidopsis thaliana plays a crucial role in cell cycle regulation.^23^ Notably, DOF3.4 expression is strongly activated in response to mechanical injury, highlighting its role in wound-induced signaling pathways.^19^ Consequently, DOF3.4 gene is a crucial component driving cell division during plant regeneration. Recent studies have shown that PAT1 subfamily proteins can promote root tip regeneration in Arabidopsis thaliana by directly activating DOF3.4.^19^
Medicago edgeworthii is a perennial herbaceous species belonging to the Fabaceae family, which mainly grows in high-altitude areas such as southeastern Xizang, southern Qinghai, and northwestern Yunnan in China.^24,25^ This species is notable not only for its high nutritional value but also for its superior adaptability to the climatic conditions of these areas, attributed to its high cold tolerance.^13^ Consequently, M. edgeworthii can be used as one of the wild high-protein pasture germplasm resources in China’s alpine grassland,^26^ and participate in the improvement, restoration and reconstruction of these fragile ecosystems. Research has shown that polyploid plants have significant advantages over diploids in terms of biomass and stress resistance. Medicago edgeworthii is naturally diploid (2n = 2x = 16) and can generate new Medicago germplasm resources through chromosome duplication.^13^ Therefore, creating a new germplasm of allopolyploid Medicago by grafting and fusing it with other Medicago species represents a promising strategy for broadening the genetic base and enhancing the agronomic traits of this species.
In this study, we identified 58 GRAS genes within the complete genome of M. edgeworthii. Comprehensive analyses were conducted to characterize their gene structure, amino acid composition, protein physicochemical properties, protein conserved motifs, chromosomal localization, and evolutionary relationships. The GRAS gene family members in M. edgeworthii were divided into nine subfamilies with GRAS proteins clustered within the same subfamily displaying conserved motifs. Here, we cloned nine putative PAT1 subfamily genes from an M. edgeworthii. A sequence alignment and phylogenetic analysis further confirmed their classification within the PAT1 subfamily, and these genes have been designated as MeGRASs. Subcellular localization experiments showed that PAT1 subfamily proteins were located in the nucleus and function as a nuclear protein. Furthermore, transient expression analysis in tobacco showed that PAT1 subfamily proteins activated the expression of MeDOF3.4 gene, indicating that PAT1 subfamily proteins might be related to promoting cell proliferation and regeneration. Collectively, these findings provided a theoretical basis for further elucidating the physiological and biochemical functions of the GRAS gene family in M. edgeworthii, thereby laying a foundation for the genetic improvement and cultivation of high-quality alfalfa varieties in the future.
Manuscript formatting
Materials and methods
Plant materials and growth conditions
All plantlets of M. edgeworthii were cultivated on Murashige & Skoog basal medium with vitamins (Phyto Technology Laboratories, M519) supplemented with 3.0% (w/v) sucrose and 0.8% (w/v) agar (pH 5.8) at temperatures of 25/18°C under a 16-h light/8-h dark photoperiod. To investigate the expression levels of PAT1 subfamily genes in different tissues of M. edgeworthii, we collected roots, stems, and leaves from 14-day-old M. edgeworthii plantlets. The collected materials were immediately frozen in liquid nitrogen and stored at −80℃ until RNA extraction.
Extraction of DNA and RNA
Total RNA was extracted according to the instructions provided with the Eastep ® Super Total RNA Extraction Kit (Promega, Madison, WI, USA). The quality of the extracted RNA was assessed using 1% agarose gel electrophoresis. Qualified RNA samples were then reverse transcribed into cDNA following the protocols of the 5X All-In-One RT MasterMix (with AccuRT Genomic DNA Removal Kit).
Identification of GRAS gene family members and analysis of basic physicochemical properties in M. edgeworthii
To identify GRAS family members in M. edgeworthii, GRAS protein sequences of M. truncatula and Arabidopsis thaliana were retrieved from the Plant Transcription Factor Database (http://planttfdb.gao-lab.org) and the Arabidopsis Information Resource (TAIR) database (http://www.Arabidopsis.org), respectively. The genome sequence data for M. edgeworthii are unpublished data generated in our laboratory. Using TBtools software,^27^ based on the known GRAS transcription factor family sequence of M. truncatula, a local BLAST search was performed on the sequence of M. edgeworthii to preliminarily determine the GRAS members in M. edgeworthii (Parameters were E < 1e^−10^, Identity > 40%). Afterwards, candidate genes were validated using the Hidden Markov Model (HMM) configuration file (login number: PF03514) obtained from the Pfam database (http://pfam.sanger.ac.uk).^28^ Additionally, the SMART tool was used to confirm the presence of conserved homologous domains within the candidate genes. Protein sequences containing GRAS typical domains (SAW, PFYRE, Leucine Heptad Repeat II (LHR II), VHIID, Leucine Heptad Repeat I (LHR I) from C-terminus to N-terminus) were screened. Genes lacking these conserved domains were excluded for further analysis, while sequences with homologous domains were retained for subsequent investigation.
Based on phylogenetic clustering with GRAS proteins of Arabidopsis and M. truncatula, the identified GRAS proteins in M. edgeworthii were designated as MeGRAS proteins. The ExPASy server^29^ (https://web.expasy.org/protparam/) was used to calculate the molecular weight (MW), theoretical isoelectric point (pI), grand average hydropathicity (GRAVY), and amino acid sequence length of the proteins. Gpos-mPLoc (http://www.csbio.sjtu.edu.cn/bioinf/Gpos-multi/.) and NPS-SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/secpred_sopma.pl) were used to predict the subcellular localization and secondary structure of MeGRAS proteins.
Phylogenetic, gene structure, and motif analysis of GRAS genes
Multiple sequence alignments of Arabidopsis thaliana, M. truncatula, and M. edgeworthii GRAS proteins were performed using MEGA 7.0.^30^ The exon-intron structures of the MeGRASs were visualized using TBtools software^27^ based on the GFF annotation file derived from the M. edgeworthii genome information. To analyze the conserved motif composition of MeGRASs, MEME (Multiple Em for Motif Elucidation) (http://meme-suite.org/index.html) was employed using default parameters.^31^ The search criteria were configured as follows: the optimal motif sequence width was set between 6 and 50 residues, with a maximum of 20 motifs identified, and other parameters were set to their default values. Each motif sequence was required to appear at least once. Additionally, conserved homologous domains encoded by MeGRAS gene were identified using MEGA 7.0^30^ and visualized with GeneDoc software.
Chromosomal location and analysis of the cis-regulatory elements in the promoter
Using the genome gff files of M. edgeworthii, TBtools software was used to perform chromosome localization analysis for each member of the GRAS gene family in M. edgeworthii. The structural features of MeGRAS proteins were analyzed using the corresponding annotation files in the genome database of M. edgeworthii. The types, quantities, and functions of cis-regulatory elements within the 2.0 kb promoter region upstream of the MeGRAS gene start codon were analyzed and visualized using the PlantCARE website (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/).^32^ Additionally, the three-dimensional structures of MeGRAS proteins were predicted using the SWISS-MODEL (https://swissmodel.expasy.org/).^33^
Quantitative real-time reverse transcriptase-polymerase chain reaction (RTqPCR)
The RT-qPCR was performed on the Applied Biosystems 7500 PCR system using Fast Start Universal SYBR Green Master Mix (ROX). The reaction mixture was prepared as follows: 10.0 μL Fast Start Universal SYBR Green Master Mix, 0.6 μL each upstream and downstream primers (10 μM) (Table S2),^34^ 2.0 μL cDNA template, 6.8 μL nuclease-free H_2_O, resulting in a total reaction volume of 20 μL. The thermal cycling conditions were as follows: an initial step at 50°C for 120 s, followed by denaturation at 95°C for 10 min, and then 40 cycles of 95°C for 15 s, 60°C for 60 s, and 72°C for 34 s. The protocol concluded with a melting curve analysis: 95°C for 15 s, 60°C for 60 s, and 95°C for 15 s. All experiments were performed in three independent biological replicates. The 2^−ΔΔCT^ method was used to calculate the relative expression levels.^35^ To determine the statistical significance of the results, one-way analysis of variance (ANOVA) was performed using IBM SPSS Statistics version 2.0 software.
Subcellular localization
Primers were designed based on the coding DNA sequence of MeGRASs in M. edgeworthii. The complementary DNA (cDNA) obtained from total RNA was then used as a template to clone the target genes using the Phanta® Max Super-Fidelity DNA Polymerase gene amplification kit (Nanjing Novozymes Biotechnology Co., Ltd). The PCR reaction conditions were as follows: an initial denaturation at 94℃ for 3 min, followed by 32 cycles of denaturation at 94℃ for 15 s, annealing at 65℃ for 30 s, and extension at 72℃ for 1 min and 45 s. A final extension was performed at 72℃ for 10 min to ensure complete product synthesis. Following gel extraction and purification of the target fragments, the amplified genes were ligated into the pRI101-GFP vector via homologous recombination. The resulting recombinant plasmids were introduced into Escherichia coli DH5α competent cells using the heat shock transformation method. The transformed cells were plated on selective media containing the appropriate antibiotic. The following day, single colonies were isolated, and a small-scale culture was carried out to extract plasmids, which were then identified by sequencing. Upon confirmation of the correct plasmid, the construct was transformed into Agrobacterium tumefaciens strain EHA105, and successful transformation was verified by PCR. Subsequently, single colonies of A. tumefaciens were selected and cultured, and the recombinant bacteria were used for transient expression assays in Nicotiana benthamiana leaves via agroinfiltration. After 72 h, GFP expression in the infiltrated leaves was observed and documented under a laser confocal scanning microscope for subcellular localization analysis.
Transient expression analysis in tobacco leaves
To generate proDOF3.4:LUC, the promoter region of DOF3.4 was amplified using the primers proDOF3.4-F (5’-GCCAGTGCCAAGCTTCAAAAGAAAAATTTTATAC-3’) and proDOF3.4-R (3’-GTCTTCCATGTCGACGTTTAGTATTTGAAAGAAG-5’). The amplified fragments were cloned into pRI101 LUC vector to generate proDOF3.4:LUC construct. These constructs were then transformed into Agrobacterium tumefaciens strain EHA105. The reporter construct was co-infiltrated into tobacco leaves along with effector constructs expressing 35S:MeGRAS14, 35S:MeGRAS24, 35S:MeGRAS38, 35S:MeGRAS41, 35S:MeGRAS42, or the corresponding empty vector control. After incubation for 3 d, the infiltrated leaves were treated with luciferin substrate and luminescence was detected using a CCD imaging system. All assays were performed with at least three independent biological replicates.
Result
Identification of GRAS family genes in M. edgeworthii
The availability of the complete M. edgeworthii genome enabled a comprehensive genome-wide identification of GRAS genes. To obtain an extensive overview of GRAS genes in M. edgeworthii, we conducted a genome-wide analysis. In the current study, we used a local BLAST search to compare the protein sequences of MtGRAS transcription factor family members from Medicago truncatula with the genome sequence of M. edgeworthii.^36^ As a result, 58 candidate MeGRAS genes were identified and named as MeGRAS1-MeGRAS58, respectively. To further investigate the physicochemical properties of the GRAS family sequence in M. edgeworthii, the online software Pfam^28^ and SMART database were used. The results obtained revealed considerable variation in the size of the full-length cDNA sequences of the MeGRAS genes, ranging from 636 bp (MeGRAS58) to 2448 bp (MeGRAS46). The full length of most GRAS proteins varied from 211 (MeGRAS58) to 815 (MeGRAS46) amino acids, with molecular weights ranging from 24.3 kDa (MeGRAS58) to 90.05 kDa (MeGRAS46). The theoretical isoelectric point (pI) ranged from 4.72 (MeGRAS50) to 9.91 (MeGRAS52). Additionally, 54 of the MeGRAS proteins had a theoretical isoelectric point (pI) <7, indicating that the majority of the MeGRAS proteins were rich in acidic amino acid residues. The calculated grand average of hydrophobicity (GRAVY) values for these proteins ranged from −0.69 (MeGRAS48) to 0.09 (MeGRAS58). Predictions of subcellular localization indicated that all members were localized in the nucleus except MeGRAS52, MeGRAS56, and MeGRAS58 (Table 1).Table 1.The physicochemical properties of MeGRAS proteins.Gene nameCDS length (bp)Protein size (aa)MW (kD)PIGRAVYMeGRAS1219072981.275.38−0.33MeGRAS2219673181.445.38−0.36MeGRAS3177959265.834.81−0.44MeGRAS4171357064.335.06−0.47MeGRAS5144948254.535.80−0.23MeGRAS6146148655.627.53−0.14MeGRAS7138646152.745.69−0.35MeGRAS8144348053.995.57−0.20MeGRAS9179159666.885.20−0.34MeGRAS1075325028.546.21−0.19MeGRAS11180059968.615.41−0.30MeGRAS12178859568.275.20−0.33MeGRAS1396031936.635.88−0.15MeGRAS14174057964.465.72−0.36MeGRAS15137745851.695.42−0.12MeGRAS16196865574.445.45−0.46MeGRAS17186962271.365.45−0.45MeGRAS18192664172.605.38−0.46MeGRAS19220573483.105.53−0.49MeGRAS20197165674.465.15−0.47MeGRAS21197165674.465.69−0.54MeGRAS22135645151.905.71−0.52MeGRAS23121840546.065.88−0.11MeGRAS24162954261.875.23−0.38MeGRAS25153651156.565.62−0.35MeGRAS26192364073.215.46−0.57MeGRAS27192364073.215.46−0.57MeGRAS28214271381.415.38−0.57MeGRAS29204668177.315.44−0.48MeGRAS30196865573.645.84−0.47MeGRAS31135945250.045.36−0.24MeGRAS32141947253.536.64−0.22MeGRAS33168956262.385.09−0.30MeGRAS34133544450.045.67−0.16MeGRAS35151550457.704.97−0.54MeGRAS36203167675.115.48−0.45MeGRAS37156652158.676.24−0.19MeGRAS38163254360.965.82−0.35MeGRAS39148549455.965.28−0.41MeGRAS40137445751.605.69−0.08MeGRAS41159953260.035.17−0.29MeGRAS42163554461.035.18−0.33MeGRAS43202867574.855.59−0.34MeGRAS44223274383.966.18−0.41MeGRAS45132641149.975.27−0.45MeGRAS46244881590.056.10−0.46MeGRAS47167155664.576.00−0.40MeGRAS48166555461.675.92−0.69MeGRAS49162654160.315.57−0.25MeGRAS50152450758.384.72−0.19MeGRAS51144348054.524.76−0.33MeGRAS52155151658.599.910.02MeGRAS5386428731.774.78−0.67MeGRAS54128142648.155.74−0.20MeGRAS55162354059.755.44−0.33MeGRAS56156051958.809.470.01MeGRAS57136545450.806.14−0.56MeGRAS5863621124.308.830.09
Secondary structure analysis showed that MeGRAS proteins were composed of four different forms of secondary structures, which are dominated by α helices and irregular coiling, while β turn angles were the least abundant across all member species (Table S1). The subcellular localization prediction indicated that all members, except for MeGRAS52, MeGRAS56 and MeGRAS58 were localized in the nucleus (Table S1).
Phylogenetic and structural analysis of MeGRAS genes
To further evaluate the evolutionary relationships of GRAS proteins across different plant species, we selected the full-length amino acid sequences of 32 AtGRAS proteins from A. thaliana, 68 MtGRAS proteins from Medicago truncatula, and 58 MeGRAS proteins from Medicago edgeworthii for multiple sequence alignment and the subsequent construction of a Neighbour-Joining phylogenetic tree. Based on the established classification of GRAS proteins in A. thaliana and M. truncatula, the 58 MeGRAS proteins from M. edgeworthii were categorized into 9 subfamilies: LISCL, SHR, PAT1, SCL3, DELLA, SCR, LS, SCL28, and HAM. The distribution of protein across these subfamilies was as follows: 11 in LISCL, 8 in SHR, 9 in PAT1, 3 in SCL3, 8 in DELLA, 3 in SCR, 2 in LS, 4 in SCL28, and 10 in HAM, respectively (Figure 1). Notably, the PAT1 subfamily members were MeGRAS3, MeGRAS4, MeGRAS9, MeGRAS10, MeGRAS14, MeGRAS24, MeGRAS38, MeGRAS41 and MeGRAS42 (Figure 1). Using the MEME suite for motif prediction, we analyzed the GRAS family genes in M. edgeworthii to discern the relationships among GRAS transcription factors and the diversity within the GRAS protein family. The analysis revealed that GRAS proteins clustered within the same subfamily share similar motifs, with nearly all GRAS members containing motifs 4, 5, and 9, underscoring their conserved nature (Supplemental Figure 1). Moreover, the C-terminus region of MeGRAS proteins contained more motifs compared to the N-terminus region. Consequently, the C-terminus of GRAS proteins were more conserved, whereas the N-terminus exhibited greater variability. This variability in the N-terminal motifs contributes to the functional diversity of GRAS genes and might also explain the distinct functions observed among different subfamilies. Figure 1.Phylogenetic tree of GRAS proteins from M. edgeworthii, M. truncatula and Arabidopsis thaliana. The unrooted phylogenetic tree was constructed by the Neighbor-Joining (NJ) method using MEGA 7.0 software incorporating 1000 bootstraps iterations. The analysis included 53 GRAS proteins, 32 from Arabidopsis thaliana, 68 from M. truncatula, and 58 from M. edgeworthii. These proteins were clustered into nine distinct subfamilies: LISCL, SHR, PAT1, SCL3, DELLA, SCR, LS, SCL28, and HAM, respectively.
To confirm whether the screened genes were members of the GRAS family, multiple sequence alignment was performed using MEGA 7.0.^30^ The alignment results showed that all the screened genes possessed one or more GRAS conserved domains at the C-terminus (Supplemental Figure 2). Specifically, these domains included SAW, PFYRE, Leucine Heptad Repeat II (LHR II), VHIID, and Leucine Heptad Repeat I (LHR I), thereby confirming that the identified genes belong to the GRAS family (Supplemental Figure S2). To further investigate the structural diversity of members of the PAT1 gene subfamily in M. edgeworthii, we identified and visualized the motif compositions of nine MeGRAS proteins using MEGA 7.0 and MEME software. A total of 20 conserved motifs were identified, labeled as motifs 1 to 20 (Figure 2a). Notably, motifs 1, 2, 3, 4, and 11 were present across most sequences, suggesting the functional similarity and conserved position among the GRAS genes in M. edgeworthii (Figure 2b). Figure 2.Phylogenetic and structural analysis of PAT1 gene subfamily. (a) Phylogenetic tree and MEME protein structure of members of PAT1 gene subfamily in M. edgeworthii. (b) Five most conserved motifs of PAT1 gene subfamily in M. edgeworthii, with the bits representing the score of the site.
Chromosomal distributions of PAT1 subfamily genes
Chromosomal localization analysis of the GRAS gene family members in M. edgeworthii was performed using TBtools software^27^ based on the available M. edgeworthii genome gff file. The 58 identified MeGRAS genes were unevenly distributed across the eight chromosomes of M. edgeworthii. Specifically, there were three genes on chromosome 6, four on chromosome 4, eight on each of chromosomes 2, 3 and 8. Additionally, 11 genes were clustered on chromosome 5, 15 on chromosome 1, and only one gene was located on chromosome 7 (Figure 3). Figure 3.Distribution and localization of MeGrass gene family members on different chromosomes. Chromosome number is indicated at the left of each chromosome. Blue represents the PAT1 subfamily genes.
Analysis of the chromosomal localization of the nine PAT1 subfamily genes according to the gene location files revealed that these genes were not distributed on chromosomes 2, 5 or 7 and were unevenly distributed across the remaining five chromosomes. Specifically, three genes (MeGRAS9, MeGRAS10, MeGRAS14) were located on chromosome 1, one gene (MeGRAS38) was located on chromosome 3, one gene (MeGRAS24) was located on chromosome 4, two genes (MeGRAS3, MeGRAS4) were located on chromosome 6, and two genes (MeGRAS41, MeGRAS42) were located on chromosome 8 (Figure 3).
Cis-acting element analyses of PAT1 subfamily genes
The cis-acting elements are crucial for the transcriptional regulation of gene expression. To further investigate the potential regulatory mechanism of the PAT1 subfamily genes in M. edgeworthii under abiotic stress, the upstream 2.0 kb sequences of the MeGRASs gene were analyzed using the PlantCARE database for detecting the presence of responsive elements. The analysis identified 14 cis-acting elements related to responses to light, drought, low-temperature stress, plant hormone signaling pathway, and defense (Figure 4a). Furthermore, cis-acting elements exhibited variability among different genes. The majority of these elements were associated with light response, such as cis-acting regulatory elements involved in light response, MYB binding sites, and gapA-CMA1. This indicated the importance of these elements in regulating gene expression, enabling plants to participate in light signal transduction pathways. These findings further indicated that GRAS transcription factors are involved in several key processes related to the growth and development of M. edgeworthii, including light signal transduction, hormone signal transduction, and stress response. Figure 4.Cis-acting element analyses of PAT1 subfamily genes. (a) Cis-acting elements of members of PAT1 gene subfamily in M. edgeworthii. Promoter sequences (−2 kb) of 9 MeGRAS genes are analyzed by PlantCARE. Rectangles with different colors indicate different cis-acting elements participating in various abiotic stress regulations. (b) Three-dimensional conformation of members of PAT1 gene subfamily in M. edgeworthii.
The function of proteins is closely related to their structural conformation. The secondary structure of PAT1 subfamily proteins, including α helices, random coils, β turns, and elongation chains, is further intricately folded through the interaction of side chain groups, resulting in the formation of a compact spherical spatial structure maintained by various secondary bonds. To further analyze the protein structure, the online tool SWISS-MODEL was used to predict the three-dimensional conformation of the PAT1 subfamily proteins. Protein tertiary structure analysis of MeGRASs, with C-terminal conserved structural domains in blue and N-terminal variable structural domains in orange, revealed that the C-terminal structural domains were predominantly α-helical with β-folding, while the N-terminal structural domains were predominantly irregularly coiled. The structure of each protein has certain similarity, which in turn dictate its functional capabilities. The results indicated that all nine members of the PAT1 gene subfamily exhibited similar three-dimensional structures (Figure 4b), suggesting that they may possess common functional characteristics.
PAT1 subfamily genes expression analysis in M. edgeworthii
To assess the expression patterns of PAT1 gene subfamily in M. edgeworthii, RT-qPCR analysis was conducted across three different tissues: roots, stems, and leaves. The results showed that all members of the PAT1 gene subfamily were expressed in the roots, stems, and leaves. However, the expression levels of each gene varied across different tissue types, indicating their broad involvement in the development of various tissues (Figure 5). Comparative analysis of gene expression across tissues revealed that MeGRAS14 had the highest expression level in leaves and shared the closest genetic relationship with Arabidopsis genes AtSCL21 and AtSCL5. Additionally, the N-terminal of MeGRAS14 contained an EAISRRDL motif, which aligned with the previously reported functions of Arabidopsis AtSCL21 and AtSCL5 in regulating the phytochrome signaling pathway.^14^ In contrast, MeGRAS24 and MeGRAS38 had the highest expression levels in stem. Phylogenetic analysis revealed that MeGRAS38 shared the closest phylogenetic relationship with AtSCL13, suggesting that MeGRAS38 mainly plays a role in the elongation zone of the hypocotyl.^15^ Furthermore, MeGRAS3, MeGRAS4, MeGRAS9, MeGRAS10, and MeGRAS42 showed the highest expression levels in roots, providing further evidence for the involvement of PAT1 subfamily genes in plant root growth and development.^20,21^ Figure 5.The relative expression levels of 9 PAT1 genes in M. edgeworthii among different tissues. The error bars indicate standard deviation (SD), and the sample size was 3. The data are analyzed by one-way ANOVA (Duncan’s test). Asterisks above columns indicate the significance of a group: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Cloning and subcellular localization analysis of partially PAT1 subfamily genes in M. edgeworthii
Determining a protein’s subcellular localization is important for elucidating its functional role within the cell. For this purpose, specific primers were designed for MeGRAS3, MeGRAS14, MeGRAS24, MeGRAS38, and MeGRAS42, for which preliminary insights into their biological functions were already available. PCR amplification was performed using cDNA from M. edgeworthii to clone these genes. The coding sequences (CDS) of the five MeGRASs were then ligated into the pRI101-GFP vector and transferred into Agrobacterium for subsequent transient expression in tobacco leaves. Confocal microscopy analysis revealed that all five MeGRAS proteins were located on the nucleus (Figure 6). Figure 6.Fluorescence map of the subcellular localization of MeGRAS3, MeGRAS14, MeGRAS24, MeGRAS38 and MeGRAS42. 35S: GFP: expression of 35S-GFP/pRI101-GFP empty vector in tobacco leaves. Scale bar = 20 μm.
The PAT1 subfamily genes activate the expression of the MeDOF3.4 gene in M. edgeworthii
The PAT1 subfamily functions as a transcription factor by binding to the promoters of DNA binding with one finger (Dof) family genes, thereby promoting cell proliferation and tissue regeneration.^23^ A previous report claimed that SCL5/SCL21/PAT1 can activate the expression of the DOF3.4 gene.^19^ To investigate whether similar transcriptional activation exists in M. edgeworthii, we constructed a proDOF3.4: LUC reporter gene line by fusing its promoter (2000 kb) to the LUC gene. Transient expression assays in tobacco leaves confirmed that MeGRAS14, MeGRAS38, MeGRAS41, and MeGRAS42 can activate the expression of the MeDOF3.4 gene (Figure 7a,b). These results suggested that the PAT1 subfamily might perform a similar function in both Arabidopsis and M. edgeworthii. Figure 7.The PAT1 subfamily genes activate the expression of the MeDOF3.4 gene in M. edgeworthii. Transient expression assays (a) and the mean of fluorescence intensity (b) confirmed that MeGRAS14, MeGRAS24, MeGRAS38, MeGRAS41, and MeGRAS42 can activate the expression of the MeDOF3.4 gene. The error bars indicate standard deviation (SD), and the sample size was 3. The data are analyzed by one-way ANOVA (Duncan’s test). Asterisks above columns indicate the significance of a group: **p < 0.01. “1” indicates “pMedof3.4:luc”; “2” indicates “pMedof3.4:luc + MeGRAS14”; “3” indicates “pMedof3.4:luc + MeGRAS24”; “4” indicates “pMedof3.4:luc + MeGRAS38”; “5” indicates “pMedof3.4:luc + MeGRAS41”; “6” indicates “pMedof3.4:luc + MeGRAS42”.
Discussion
GRAS, as unique transcription factor in plants, plays an important regulatory role in several biological processes such as plant growth and development, hormone and light signaling, as well as abiotic stress response.^12^ However, to date, no comprehensive analysis of this gene family has been conducted in Medicago edgeworthii.
This study identified and characterized 58 members of the MeGRAS gene family from the entire genome of M. edgeworthii. All MeGRAS proteins showed significant structural diversity, indicating a high level of complexity of this gene family. In this study, the amino acid length of the proteins encoded by the GRAS gene family members in M. edgeworthii varied from 211 to 815 amino acids, which was comparable to the variability observed in other plant species such as maize,^37^ soybean,^38^ and Eucalyptus grandis.^39^ Similarly, the isoelectric points (pI) of these proteins varied from 4.72 to 9.91, with 54 proteins having an isoelectric point below 7 and 4 proteins having an isoelectric point above 7, suggesting a predominance of acidic proteins. These results were consistent with those reported for soybean^38^ and sweet potato,^40^ indicating that the physicochemical properties of GRAS gene family members were conserved across different species. The GRAS genes of M. edgeworthii were categorized into nine subfamilies. GRAS proteins clustered within the same subfamily had similar motifs, suggesting that members of the same subfamily may perform similar functions.
Due to the close relationship between the structure and properties of proteins and their functions, this study analyzed the protein properties and structural characteristics of the GRAS gene family in M. edgeworthii. By comparing the full-length sequence of amino acids, it was found that the C-terminus of the GRAS protein amino acid sequence in M. edgeworthii had high conservation, characterized by the presence of five conserved motifs: LHRI/II, VHIID, PYRE, and SAW. In contrast, the N-terminal sequences of these amino acid sequences had significant differences, and the predicted results of the protein’s tertiary structure were also consistent. Subcellular localization software predicted the localization of GRAS protein in the nucleus. Subcellular localization experiments in tobacco leaves revealed the, nuclear localization, which was consistent with the results of the analysis software.
Members of the Arabidopsis PAT1 gene subfamily are involved in plant life activities such as defense against stress, elongation of hypocotyl, light signal response pathways,^11^ and cell fusion reactions.^20^ Structural analysis and examination of conserved protein domains of the M. edgeworthii PAT1 gene subfamily revealed that among the 25 predicted cis-acting elements identified in the 9 PAT1 gene subfamily members, 14 were related to plant hormones, light, drought stress, low temperature, defense, and stress responses. These findings were consistent with existing research reports. The MeGRAS3, MeGRAS24, and MeGRAS14 genes had high expression levels in roots, stems, and leaves, respectively. It was speculated that these genes mainly participate in growth and development of roots and hypocotyls, as well as to participate in light response signal transduction pathways. Previous studies have shown that the PAT1 gene subfamily is a conserved regulator of graft healing and tissue regeneration,^20^ while the DOF3.4 gene is a key factor driving cell division during plant regeneration.^19^ Notably, Arabidopsis SCL5/SCL21/PAT1 have been shown to activate the expression of DOF3.4 gene, which promotes cell division and tissue regeneration.^19^ In this study, tobacco transient expression experiments showed that several PAT1 subfamily proteins in M. edgeworthii can also activate the expression of MeDOF3.4 gene. Further explanation suggests that the PAT1 subfamily may have similar functions in Arabidopsis and M. edgeworthii.
In conclusion, using TBtools software,^27^ we discovered and characterized nine members of the M. edgeworthii PAT1 gene subfamily. A comprehensive analysis was conducted into their phylogenetic relationships, gene structures, expression profiles across various tissues, and subcellular localizations. Our results suggested that these nine PAT1 subfamily genes may play a role in defense against stress response, hypocotyl growth and development, light signal response pathways, and cell fusion response. Overall, the genomic identification and bioinformatics analysis of the M. edgeworthii PAT1 gene subfamily provide a reference for further elucidation of the functions of MeGRAS family genes in M. edgeworthii, and provide promising candidate genes for the molecular breeding and genetic improvement of M. edgeworthii.
Supplementary Material
Supplemental Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Silverstone AL, Ciampaglio CN, Sun T-P. The Arabidopsis RGA gene encodes a transcriptional regulator repressing the gibberellin signal transduction pathway. Plant Cell. 1998;10(2):155–12. doi: 10.1105/tpc.10.2.155.9490740 PMC 143987 · doi ↗ · pubmed ↗
- 2Bolle C. The role of GRAS proteins in plant signal transduction and development. Planta. 2004;218(5):683–692. doi: 10.1007/s 00425-004-1203-z.14760535 · doi ↗ · pubmed ↗
- 3Di Laurenzio L, Wysocka-Diller J, Malamy JE, Pysh L, Helariutta Y, Freshour G, Hahn MG, Feldmann KA, Benfey PN. The SCARECROW gene regulates an asymmetric cell division that is essential for generating the radial organization of the Arabidopsis root. Cell. 1996;86(3):423–433. doi: 10.1016/S 0092-8674(00)80115-4.8756724 · doi ↗ · pubmed ↗
- 4Peng J, Carol P, Fau -Richards DE, Richards de Fau - King KE, King Ke Fau - Cowling RJ, Cowling Rj Fau - Murphy GP, Murphy Gp Fau - Harberd NP. The Arabidopsis GAI gene defines a signaling pathway that negatively regulates gibberellin responses. Genes Devel. 1997;11(23):3194–3205. doi: 10.1101/gad.11.23.3194.9389651 PMC 316750 · doi ↗ · pubmed ↗
- 5Pysh LD, Wysocka-Diller JW, Camilleri C, Bouchez D, Benfey PN. The GRAS gene family in Arabidopsis: sequence characterization and basic expression analysis of the SCARECROW-LIKE genes. Plant J. 1999;18(1):111–119. doi: 10.1046/j.1365-313X.1999.00431.x.10341448 · doi ↗ · pubmed ↗
- 6Ni L, Wang Z, Liu X, Wu S, Hua J, Liu L, Yin Y, Li H, Gu C. Genome-wide study of the GRAS gene family in Hibiscus hamabo Sieb. et Zucc and analysis of Hh GRAS 14-induced drought and salt stress tolerance in Arabidopsis. Plant Sci. 2022;319:111260. doi: 10.1016/j.plantsci.2022.111260.35487668 · doi ↗ · pubmed ↗
- 7Fan Y, Wan X, Zhang X, Zhang J, Zheng C, Yang Q, Yang L, Li X, Feng L, Zou L, et al. GRAS gene family in rye (Secale cereale L.): genome-wide identification, phylogeny, evolutionary expansion and expression analyses. BMC Plant Biol. 2024;24(1):46. doi: 10.1186/s 12870-023-04674-1.38216860 PMC 10787399 · doi ↗ · pubmed ↗
- 8Hirsch S, Oldroyd GED. GRAS-domain transcription factors that regulate plant development. Plant Signal & Behav. 2009;4:698–700. doi: 10.4161/psb.4.8.9176.PMC 280137919820314 · doi ↗ · pubmed ↗