De novo retrotransposon insertion into the FGFR1 gene in a boy with congenital hypogonadotropic hypogonadism: a case report
Kentaro Sawano, Keisuke Nagasaki, Erina Suzuki, Yasuko Ogiwara, Ikuko Kageyama, Maki Fukami, Yoko Kuroki

TL;DR
A 6-year-old boy with a rare hormonal disorder had a new genetic mutation in the FGFR1 gene caused by a retrotransposon insertion.
Contribution
This is the first reported case of CHH caused by a de novo retrotransposon insertion in the FGFR1 gene.
Findings
A de novo 333-bp Alu insertion in exon 9 of FGFR1 caused a frameshift and premature termination.
The insertion showed hallmarks of retrotransposition, including target site duplication and a poly-A tail.
The case highlights the need for long-read sequencing in CHH patients with negative short-read and array results.
Abstract
Congenital hypogonadotropic hypogonadism (CHH) is a rare endocrine disorder characterized by gonadal dysfunction attributed to impaired gonadotropin secretion. CHH is associated with approximately 60 genes including FGFR1. Nevertheless, the nucleotide variants of these genes are only related to less than half of the cases. Herein, we report a case of CHH caused by a novel mechanism. A 6-year-old boy presented with hypomasculinized genitalia, hyposmia, and syndactyly. Endocrine examinations showed impaired gonadotropin secretion. Short-read next-generation sequencing (NGS) identified the absence of mutations in the major causative genes for CHH. However, it detected an accumulation of discordant and split reads in a genomic region within the FGFR1 gene. Array-based comparative genomic hybridization did not detect copy-number abnormalities. Targeted long-read NGS and Sanger sequencing…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
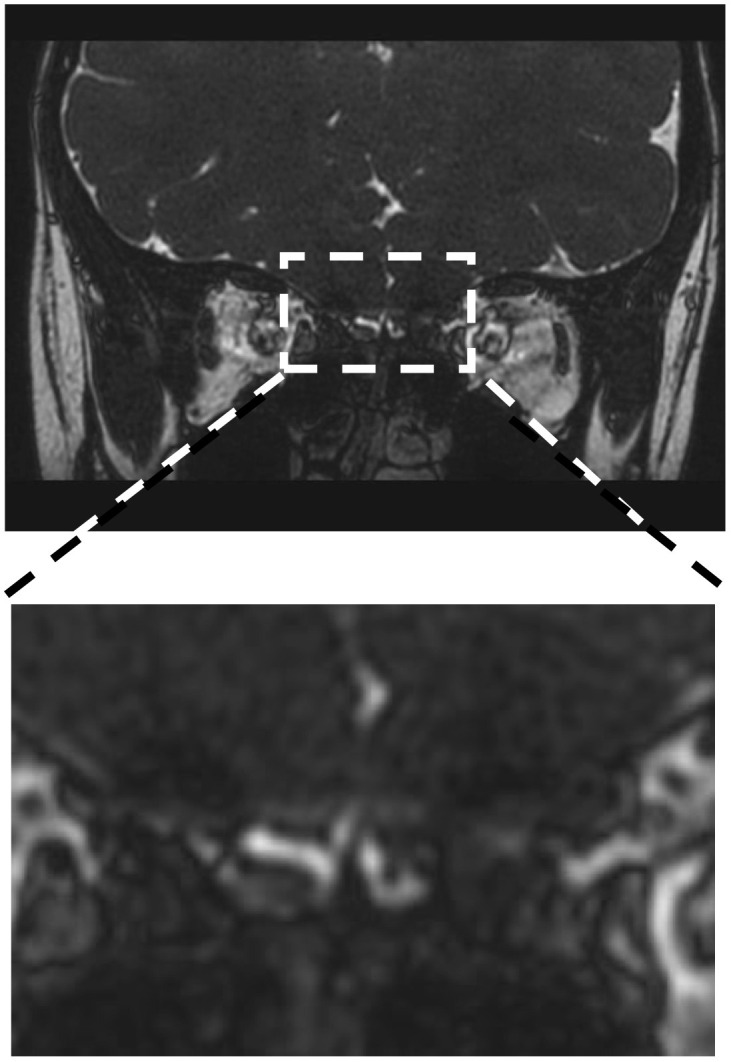 Figure 1
Figure 1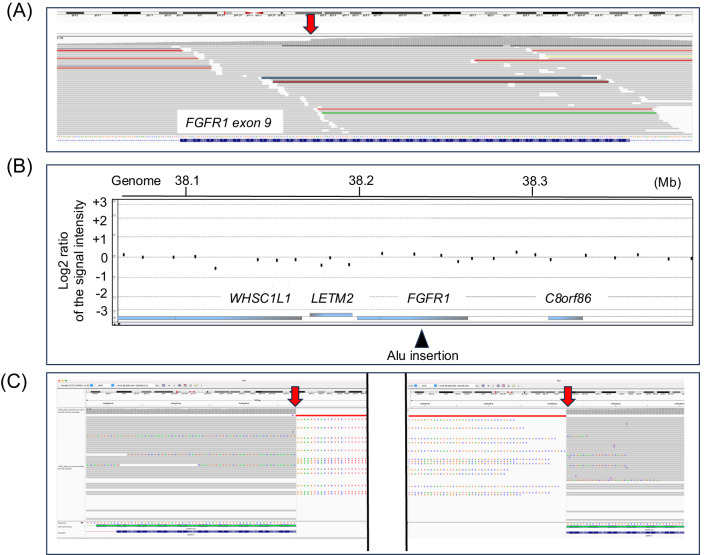 Figure 2
Figure 2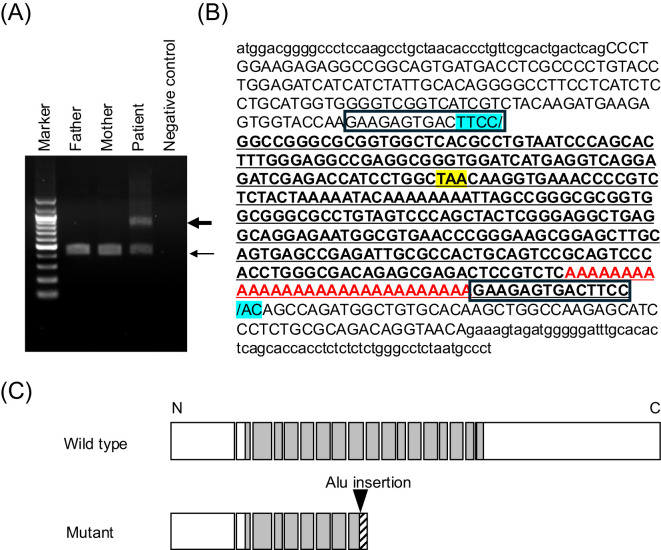 Figure 3
Figure 3| Age at examination | Luteinizing hormone level (IU/L) | Follicle-stimulating hormone level (IU/L) | Testosterone level (nmol/L) | |||
|---|---|---|---|---|---|---|
| Patient | Reference intervals | Patient | Reference intervals | Patient | Reference intervals | |
| 0 years, 3 months | <0.2 | 0.62–4.08 | <1.0 | 0.41–3.02 | 0.13 | 0.69–7.60 |
| 0 years, 6 months | <0.2 | 0.10–8.06 | 1.0 | 0.19–3.63 | <0.12 | 0.42–5.77 |
| 1 years, 7 months | <0.2 | 0.02–0.15 | <1.0 | 0.38–1.11 | <0.12 | <0.35 |
| 2 years, 8 months | <0.2 | 0.02–0.15 | <1.0 | 0.38–1.11 | 0.12 | <0.35 |
| 3 years, 8 months | <0.2 | 0.02–0.15 | <1.0 | 0.38–1.11 | <0.12 | <0.35 |
| 5 years, 4 months | <0.2 | 0.02–0.15 | <1.0 | 0.38–1.11 | 0.22 | <0.35 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCongenital Anomalies and Fetal Surgery · Genetics and Neurodevelopmental Disorders · Neurogenetic and Muscular Disorders Research
Introduction
1
Congenital hypogonadotropic hypogonadism (CHH) is a rare endocrine disorder characterized by gonadal dysfunction attributed to the impaired secretion of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the pituitary gland (1). CHH typically causes hypomasculinized external genitalia in male neonates and delayed puberty in children of both sexes (1). Patients with CHH frequently exhibit additional clinical features such as anosmia/hyposmia and skeletal abnormalities (2–4).
CHH is a genetically heterogeneous condition. This disorder is associated with approximately 60 genes, including FGFR1 (5–7). The most common pathogenic variants in these genes were single nucleotide substitutions and small indels. Meanwhile, the structural variants, such as deletions, insertions, and translocations, accounted for a small percentage of the cases (1). Based on a previous study, systematic mutation screening using short-read next-generation sequencing (NGS) and genome-wide copy-number analysis with array-based comparative genomic hybridization (CGH) identified pathogenic variants in less than half of patients with CHH (8). Therefore, unrecognized genetic abnormalities play a significant role in the development of CHH. In this context, de novo retrotransposon insertion into the genome is considered a rare cause of various congenital disorders (9). Nevertheless, it is not linked to CHH. Herein, we report the first case of CHH caused by retrotransposon insertion.
Case description
2
The patient was a 6-year-old boy. Table 1 shows the clinical characteristics of the patient. He was the first child of a nonconsanguineous Japanese couple. His parents had normal clinical characteristics. The patient was born at 38 weeks of gestation via cesarean section because his mother experienced hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome. His birth weight and height were 1,668 (−3.8 standard deviation [SD]) g and 43 (−2.4 SD) cm, respectively. At birth, he presented with micropenis (stretched penile length: 20 mm), bilateral cryptorchism, and 3/4 syndactyly of the right foot. Endocrine examinations at 3 months of age during minipuberty revealed low LH, FSH, and testosterone levels. The gonadotropin-releasing hormone stimulation test performed at 7 months of age revealed basal/peak LH and FSH levels at <0.2/2.3 and 1.1/24.8 IU/L, respectively. The peak LH and FSH levels were within the normal range, which indicated hypogonadotropic hypogonadism. Magnetic resonance imaging showed hypoplasia of the left olfactory groove and olfactory bulb (Figure 1). Thus, the patient was clinically diagnosed with CHH. He underwent orchidopexy at 2.1 years of age. Repeated endocrine examinations during childhood invariably showed low blood gonadotropin and testosterone levels. At 5.4 years of age, he had standard height and weight. His penile size was 35 (−1.8 SD) mm. The testes, with a volume of 1.0 mL, were palpable in the scrotum. Olfactory testing with a T&T olfactogram revealed mild olfactory loss. In particular, the right olfactory bulb had a normal function. However, olfactory perception was not detected on the left side. This finding was in accordance with the magnetic resonance imaging results.
Clinical findings of the patient. Magnetic resonance imaging showed hypoplasia of the left olfactory groove and olfactory bulb.
Molecular analyses
3
Genomic DNA samples were obtained from the peripheral leukocytes of the patient and his parents. The DNA sample of the patient was subjected to mutation screening of the major causative genes for CHH (ANOS1, CHD7, FGF8, FGFR1, GNRH1, GNRHR, IGSF1, KISS1, KISS1R, MKRN3, PROK2, PROKR2, SOX10, TAC3, TACR3, and WDR11) (5–7). Short-read NGS was performed at the Kazusa DNA Research Institute (Chiba, Japan). The variants were analyzed, as described in a previous study (14). Results showed the absence of pathogenic nucleotide variants in the coding region of the 16 genes. However, the graphic visualization of the NGS data using the Integrative Genomics Viewer (https://igv.org) revealed an accumulation of discordant reads and split reads in a genomic region within the FGFR1 gene (Figure 2A). These results indicated the presence of a structural variant in this region. Thus, CGH analysis was performed using a catalog array (Agilent Technologies, CA, the USA). However, copy-number variations were not identified in the genome of the patient (Figure 2B).
Representative results of the molecular analyses. (A) The results of short-read next-generation sequencing (NGS). Several discordant and split reads were identified in a genomic region within the FGFR1 gene. (B) The results of array-based comparative genomic hybridization. No copy-number variations were detected in the genomic region around the FGFR1 gene. (C) The results of the long-read NGS. The findings suggested a 333-bp insertion in exon 9. The arrowheads depict the insertion site.
Then, targeted long-read sequencing was conducted using the patient’s DNA sample (Oxford Nanopore Technologies, Oxford, England). An approximately 100-kb genomic interval harboring FGFR1 (T2T-CHM13/hs1, chr8:38,658,107–38,758,601) was subjected to adaptive sampling, as reported in a previous study (15, 16). Results showed a heterozygous insertion of an approximately 330-bp DNA fragment in exon 9 of the FGFR1 gene (Figure 2C). Genomic region encompassing the insertion was PCR-amplified, and the PCR products were sequenced (Figure 3A). The insertion was found to be a 333-bp fragment (Figure 3B). A sequence similarity search using RepeatMasker (https://www.repeatmasker.org) revealed that the inserted sequence was an Alu element. The inserted sequence had known hallmarks of retrotransposition such as a target site duplication, an endonuclease cleavage site-like motif (TTCC/AC) at the breakpoints, and a poly-A tail (Figure 3B) (17, 18). This insertion has not been reported in patients with CHH and is absent from the gnomAD SV database (https://gnomad.broadinstitute.org/). This insertion was predicted to cause a frameshift from the 409th codon of the FGFR1 gene and result in premature termination at the 440th codon (p. His409fsTer31) (Figure 3C). PCR using the DNA samples of the patient’s parents yielded no products encompassing the insertion. Therefore, the insertion of the patient was a de novo variant (Figure 3A).
Characterization of the Alu insertion. (A) PCR analysis of exon 9 of the FGFR1 gene in the patient and his parents. The thin arrow indicates normal PCR products (422 bp). Meanwhile, the thick arrow denotes aberrant PCR products with the Alu insertion (755 bp). The aberrant PCR products were only obtained from the patient. (B) The sequences flanking the Alu insertion. Nucleotides in exon 9 are written in uppercase. The inserted sequence is underlined. The GAAGAGTGACTTCC sequences represent the target site duplication characteristic for retrotransposition. The inserted sequence containing the endonuclease cleavage site-like motif (TTCC/AC) is marked in light blue and the poly-A tail in red. (C) The predicted structure of the mutant protein. The Alu insertion was predicted to cause a frameshift and result in premature termination (p. His409fsTer31).
Discussion
4
In the current case, an unreported 333-bp insertion in the FGFR1 gene was found in a boy with congenital hypogonadotropic hypogonadism. The patient exhibited CHH, hyposmia, and syndactyly, which are common features of FGFR1 haploinsufficiency (19, 20). The patient presented with severe intrauterine growth failure, which is rare in patients with FGFR1 abnormalities. However, this phenotype can be attributed to HELLP syndrome (21, 22). Based on these results, the insertion of the patient destroyed the function of the FGFR1 gene on one allele. Indeed, the mutated mRNA satisfied the condition of nonsense-mediated mRNA decay (23). This case emphasizes the importance of submicroscopic structural variants in the etiology of CHH.
In this case, the insertion was an Alu element. This insertion was a de novo variant, and it exhibited several hallmarks of retrotransposition such as target site duplication of 8–18 bp, the endonuclease cleavage site-like motif, and a poly-A tail. Alu is a primate-specific retrotransposon that accounts for approximately 10% of the human genome (24, 25). Retrotransposition of Alu and other repetitive elements is a common mechanism during gametogenesis or early embryogenesis. The rate of germline Alu retrotransposition is as high as 1 in 40 births (26). Retrotransposon insertions in the germline predominantly occur in intergenic or heterochromatin regions and usually permit a normal phenotype (26). However, these insertions can disrupt the coding regions or cis-regulatory elements, thereby causing genetic disorders (27). To date, patients with various monogenic disorders have presented with retrotransposon insertions into coding regions or regulatory elements (9, 28). To the best of our knowledge, this is the first case of CHH caused by retrotransposon insertion. Importantly, the insertion was not identified by short-read NGS. Nevertheless, the accumulation of split and discordant reads indicated the presence of a structural variant in the FGFR1 gene. Further, the insertion was overlooked by the array-based CGH. This can be explained by the fact that the probes of the custom CGH arrays are designed primarily in single-copy regions. Hence, retrotransposons may be hidden in other patients with CHH of unknown etiology.
In conclusion, the study results broadened the genetic basis of CHH that includes retrotransposition insertion. Based on this case, patients with CHH who have negative results for short-read NGS and array-based CGH require additional genomic analyses.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Young J Xu C Papadakis GE Acierno JS Maione L Hietamäki J. Clinical management of congenital hypogonadotropic hypogonadism. Endocr Rev. (2019) 40:669–710. doi: 10.1210/er.2018-00116 30698671 · doi ↗ · pubmed ↗
- 2Legouis R Hardelin JP Levilliers J Claverie JM Compain S Wunderle V. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell. (1991) 67:423–35. doi: 10.1016/0092-8674(91)90193-3 1913827 · doi ↗ · pubmed ↗
- 3Franco B Guioli S Pragliola A Incerti B Bardoni B Tonlorenzi R. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. (1991) 353:529–36. doi: 10.1038/353529 a 0 1922361 · doi ↗ · pubmed ↗
- 4Palmert MR Dunkel L. Clinical practice. Delayed puberty. N Engl J Med. (2012) 366:443–53. doi: 10.1056/NEJ Mcp 1109290 22296078 · doi ↗ · pubmed ↗
- 5Lima Amato LG Latronico AC Gontijo Silveira LF. Molecular and genetic aspects of congenital isolated hypogonadotropic hypogonadism. Endocrinol Metab Clin North Am. (2017) 46:283–303. doi: 10.1016/j.ecl.2017.01.010 28476224 · doi ↗ · pubmed ↗
- 6Butz H NyírőG Kurucz PA LikóI Patócs A. Molecular genetic diagnostics of hypogonadotropic hypogonadism: from panel design towards result interpretation in clinical practice. Hum Genet. (2021) 140:113–34. doi: 10.1007/s 00439-020-02148-0 PMC 786483932222824 · doi ↗ · pubmed ↗
- 7Al Sayed Y Howard SR. Panel testing for the molecular genetic diagnosis of congenital hypogonadotropic hypogonadism - a clinical perspective. Eur J Hum Genet. (2023) 31:387–94. doi: 10.1038/s 41431-022-01261-0 PMC 1013325036517585 · doi ↗ · pubmed ↗
- 8Izumi Y Suzuki E Kanzaki S Yatsuga S Kinjo S Igarashi M. Genome-wide copy number analysis and systematic mutation screening in 58 patients with hypogonadotropic hypogonadism. Fertil Steril. (2014) 102:1130–6.e 3. doi: 10.1016/j.fertnstert.2014.06.017 25064402 · doi ↗ · pubmed ↗