Tuning the Inter-Chromophore Electronic Coupling in Perylene Diimide Dimers with Rigid Covalent Linkers
Guo Yu, Yixuan Gao, Yonghang Li, Yiran Tian, Xiaoyu Zhang, Yandong Han, Jinsheng Song, Wensheng Yang, Xiaonan Ma

TL;DR
This paper presents a new strategy for tuning electronic coupling in perylene diimide dimers using rigid covalent linkers, which could improve organic optoelectronic materials.
Contribution
A new molecular design strategy for tuning inter-chromophore electronic coupling using rigid covalent linkers is proposed.
Findings
Rigid linking cores ensure minimal structural relaxation between S0 and S1 states.
Saddle-shaped linkers allow tuning of dihedral and slipping angles to control electronic coupling.
The method enables |JCoul| values ranging from 0 to 1000 cm−1.
Abstract
The organic multi-chromophore system has been increasingly attractive due to the potential optoelectronic applications. The inter-chromophore electronic coupling (EC), i.e., JCoul and JCT, plays a critical role in determining the relaxation path of the excited state. However, the molecular designing strategy for effective tuning of inter-chromophore EC is still challenging. In this computational work, we designed a series of perylene diimides (PDI) covalent dimers with rigid linking cores containing thiophene (Th) or phenyl (Ph) fragments and performed corresponding theoretical investigation to analyze the inter-PDI electronic coupling. Vibrational analysis indicated that the minimized excited state structural relaxation (ES-SR) can ensure the rigid inter-PDI geometry pre-defined by the topological characteristic of linking cores, leading to comparable |JCoul| on S0 and S1 states. The…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
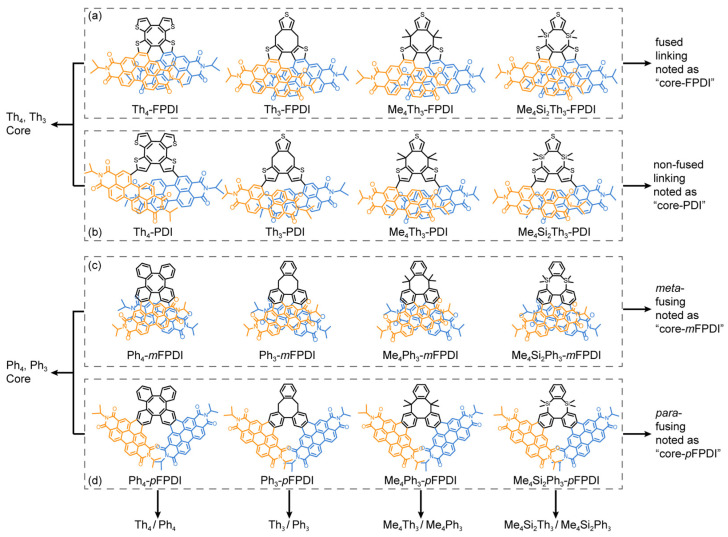 Figure 1
Figure 1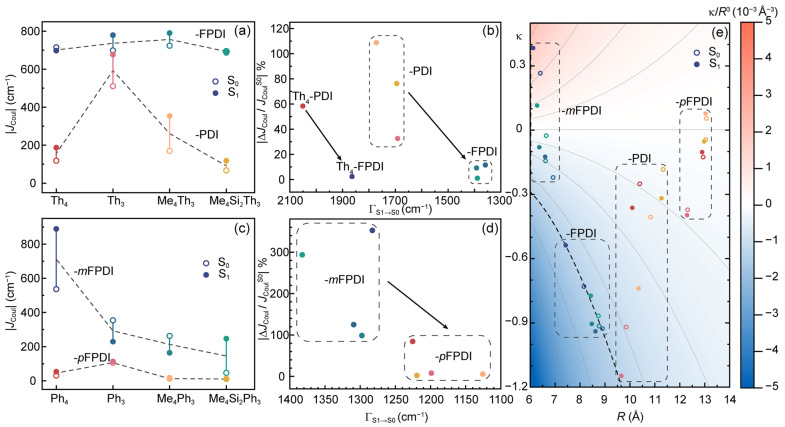 Figure 2
Figure 2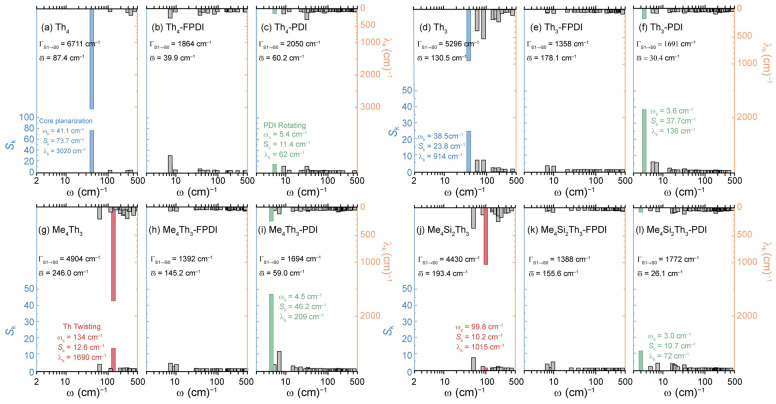 Figure 3
Figure 3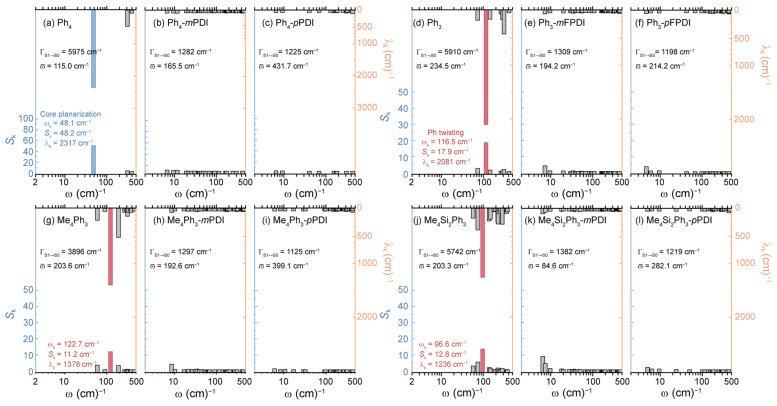 Figure 4
Figure 4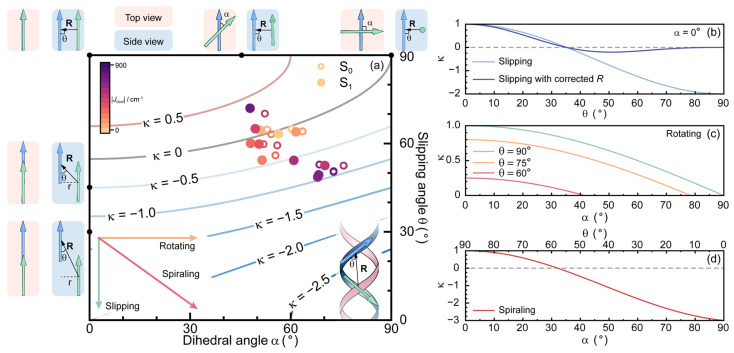 Figure 5
Figure 5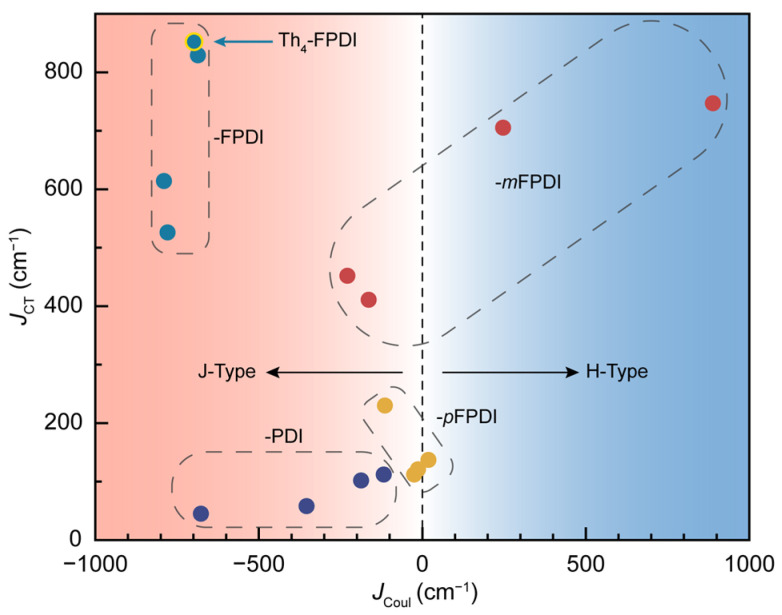 Figure 6
Figure 6- —National Key R&D Program of China
- —Natural Science Foundation of Henan Province
- —Program sponsored by Henan Province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLuminescence and Fluorescent Materials · Organic Electronics and Photovoltaics · Photoreceptor and optogenetics research
1. Introduction
Inter-chromophore electronic coupling (EC) plays a key role in determining the excited state behavior of multi-chromophore systems and optoelectronic performance of organic semiconductors with ordered molecular packing [1,2,3,4,5,6,7]. In the weak coupling regime, photoinduced dynamics of chromophores can be dominated by the locally excited (LE) state. With enhanced inter-chromophore EC combining with further condition such as energy matching, the initially populated LE state may be obtained through charge transfer (CT) [8,9,10,11,12], symmetry-breaking charge separation (SB-CS) [13,14,15,16,17,18], singlet fission (SF) [19,20,21,22,23,24], and even formation of excimer/exciplex state [25,26,27,28,29,30,31]. Recently, photo-induced SB-CS [32,33,34] and SF [35,36,37] in covalent oligomers received great attention due to potential application in improving power conversion efficiency (PCE) of organic photovoltaics (OPV) [38,39,40,41,42,43]. For enabling efficient SB-CS and SF in organic multi-chromophore systems, a key task would be smart molecular designing to achieve delicate inter-chromophore EC [20,23,44], which remains challenging.
The inter-chromophore EC has been intensively discussed in the framework of the exciton theory pioneered by Davydov [45], Kasha [46,47,48], and Spano [49,50,51,52]. Briefly, inter-chromophore EC (J) can be described as the sum of Columbic coupling (JCoul) and CT-mediated super-exchange (JCT), i.e., J = JCoul + JCT, [4,51,52] in which JCoul and JCT correspond to the long-range dipole-dipole interaction and frontier orbital overlapping in short-range, respectively. In particular, JCoul can be tunable by molecular designing as it is associated with inter-chromophore geometry, i.e., inter-chromophore distance (R), dihedral angle (α), and slipping angle (θ), which has been described by [45,48,53,54].
The definition of α and θ angles can be seen in the illustration of Figure S1. In comparison, JCT is a short-range interaction originating from frontier orbital overlapping and associated with electron (te) and hole (th) transfer integral, as well as adiabatic energy of CT (ECT) and the lowest-lying state (ES1) [51,55,56]. Please note that JCT can also be greatly affected by geometric parameters, such as inter-chromophore slipping in a co-facial dimer [51], but it relies on a short inter-chromophore distance (typically a few angstroms).
Unlike excimer/exciplex formation, which relies on strong EC [30,57,58], efficient SB-CS and SF require moderate EC, corresponding to the “degraded” structure rather than fully stacked inter-chromophore geometry, i.e., inter-chromophore geometry should be deviated from the co-facial stacking geometry to reduce EC [59,60], which is often accompanied by changes of inter-chromophore dihedral (α) and slip angles (θ). Therefore, enormous efforts in molecular designing have been made to achieve the “degraded” inter-chromophore geometry. For instance, taking advantage of excited state structural relaxation (S_1_/S_0_ ES-SR), multi-chromophore systems with flexible linkers have been widely employed for facilitating efficient SB-CS and SF [59,60,61,62,63,64]. However, considerable S_1_/S_0_ ES-SR also leads to unpredictable EC in the sense of molecular designing. Recently, we demonstrated a new molecular designing strategy for this issue [65]. By using a saddle-shaped cyclooctatetrathiophene (COTh) core and fused linking with perylene diimides (PDI), optimized inter-PDI EC (|JCoul| ≈ 700 cm^−1^) was achieved, by which efficient inter-PDI SF (τ_SF_ = ~150 ps and ~150% triplet yield) was facilitated. Considering the rigid COTh structure and fused linking with PDIs, the inter-PDI geometry that can greatly affect JCoul can be defined by the topological characteristics of COTh. In this sense, we should be able to achieve fine-tuning of inter-PDI EC (JCoul) only by modifying the structure of the rigid linking core, by which molecular designing of multi-chromophore systems can be simplified, i.e., only the geometry of the rigid core should be considered to facilitate the expected EC.
In this work, we further extended our molecular designing strategy for multi-chromophore systems. By using different rigid linkers with thiophene (Th) or phenyl (Ph) fragments, a series of PDI dimers was theoretically designed, leading to a broad tuning range of inter-PDI EC (0 < |JCoul| < 1000 cm^−1^). The vibrational analysis indicated that the S_1_/S_0_ ES-SR of rigid linking cores can be further suppressed by linking with PDI units, leading to comparable |JCoul| on S_0_ and S_1_ states, for which inter-PDI geometry was pre-defined by rigid linking cores. Considering the saddle-shaped feature of involved linking cores, our strategy enabled collaborative tuning of inter-PDI dihedral (α) and slipping (θ) angles, which can greatly neutralize the extension of inter-PDI distance (R) and lead to effective tuning of inter-PDI |JCoul|. Our work provides a new molecular designing strategy for fine-tuning of inter-chromophore EC for organic chromophores like PDI without disturbing S_1_/S_0_ ES-SR, which might be useful for future applications in organic optoelectronics.
2. Results and Discussion
2.1. Molecular Designing
In the present work, two structural features were considered for molecular designing of PDI dimers: (1) structure of rigid linking core; (2) linking manner between rigid core and PDI units. As illustrated in Figure 1a, we first modified the COTh (noted as Th_4_ in this work) core to the Th_3_ core, in which the structural symmetry can be reduced. The Me_4_Th_3_ and Me_4_Si_2_Th_3_ cores were subsequently designed to further enhance the structural rigidity. Two PDI units can be further introduced on each Th-based core with fused (rigid, Figure 1a) and non-fused (flexible, Figure 1b) linking, respectively. In comparison between PDI dimers illustrated in Figure 1a,b, we can further verify the key role of pre-defining inter-PDI geometry by rigid linking cores. Similarly, we replaced all Th units on linking the core with phenyl units, leading to corresponding cores of Ph_4_, Ph_3_, Me_4_Ph_3_, and Me_4_Si_2_Ph_3_. For each Ph-based core, PDI units can be fused to the linking core on meta- (Figure 1c) and para-position (Figure 1d) of the center cyclooctatetraene, leading to PDI dimers with variable EC due to changed inter-PDI distance (R), dihedral (α), and slipping angle (θ).
The optimized geometry on S_0_ and S_1_ states of the involved linking cores was calculated by using DFT and TDDFT approaches on B3LYP/6-311g** level, respectively. We employed the twisting dihedral angle (δ, as illustrated in Figure S2) for describing the saddle-shaped geometric feature of linking cores. As summarized in Table 1, all investigated Th- and Ph-based cores exhibited greatly reduced δ_S1_ than δ_S0_, corresponding to pronounced S_1_/S_0_ ES-SR. However, we found that S_1_/S_0_ ES-SR of cores can be greatly suppressed by linking with PDI units, leading to comparably twisted (60 to 70°) δ_S1_ and δ_S0_ angles. The calculated root of the mean of squared displacement (RMSD_S1/S0_) between S_1_ and S_0_ state also indicated less pronounced S_1_/S_0_ ES-SR of linking cores in PDI dimers, which might be explained by two aspects of factor [66,67]. Firstly, the steric effect induced by PDI units may suppress S_1_/S_0_ ES-SR of linking cores, which can be verified by lower core RMSD_S1/S0_ in non-fused dimers (i.e., Th_3_-PDI, etc.) than in corresponding fused dimers (i.e., Th_3_-FPDI, etc.). Meanwhile, considering the higher S_0_→S_1_ energy gap of the investigated linking cores (>3 eV) than the PDI unit (<2.5 eV), the cores might not be greatly involved in the S_0_→S_1_ transition of the corresponding PDI dimers. The linking cores with minimized S_1_/S_0_ ES-SR in PDI dimers ensured that the inter-PDI geometry can be pre-defined by rigid cores for tuning the inter-PDI JCoul.
2.2. Electronic Coupling
As described above, the inter-PDI EC in PDI dimers includes contributions of JCoul and JCT, in which JCoul is highly dependent on the inter-PDI geometry, i.e., inter-PDI distance (R), dihedral angle (α), and slipping angle (θ). Meanwhile, JCT is a short-range interaction that is dependent on overlapping and alignment of frontier orbitals. The term of (cosα − 3cos^2^θ) involved in Equation (1) of JCoul can usually be defined as an angular factor (κ) which determines the positive and negative values of JCoul. In exciton theory, JCoul < 0 and JCoul > 0 imply the J- (head-to-tail) and H-type (side-by-side) interaction among chromophores.
In contrast, JCT is a sort of short-range interaction associated with frontier orbital overlapping, which is considerably dependent on inter-PDI geometry, only with short inter-PDI distance (typically a few angstroms). The JCoul (Equation (1)) of all designed PDI dimers with S_0_ and S_1_ geometries was calculated together with corresponding JCT (Equation (2)), leading to values of κ_S0_, κ_S1_, JCoul^S0^, JCoul^S1^, and JCT summarized in Table 2. The geometric parameters of PDI dimers (α, θ, and R) for calculating JCoul on S_0_ and S_1_ states can be seen in Table S1. The corresponding parameters for calculating inter-PDI JCT were obtained by using the Multiwfn program [68,69], and are listed in Table S2.
As shown in Figure S3a, calculated JCT of PDI dimers exhibited a jump-like dependence on inter-PDI distance, i.e., JCT > 400 cm^−1^ in the area of R = 6–9 Å and JCT < 200 cm^−1^ in the area of R = 9–12 Å, which is consistent with the short-range nature of JCT. Meanwhile, dependence of JCT on α and θ angles can be barely observed (Figure S3b,c), which is dramatically different from the case of slipping perylene dimers with 3.5 Å stacking distance [51]. Please note that the positive/negative sign of JCT is highly dependent on the sign of th and te. However, a sign of th and te is determined by the phase assignment of the frontier orbital under a specific symmetry operation [52]. To avoid redundant discussion, we treated th and te with the definition by Wasielewski et al. [3], in which th and te can be defined with a different sign as moving a hole to one direction corresponds to moving an electron to the reverse direction.
We further discuss the calculated inter-PDI JCoul that is highly dependent on inter-PDI geometry in designed covalent dimers. As shown in Figure 2a, PDI dimers with fused linking to Th-based cores (Th-FPDIs) exhibited large α angle (60–70°) and slightly slipping (θ ≈ 50°) inter-PDI geometry on the S_1_ state, corresponding to a degraded stacking geometry with comparably strong inter-PDI EC (|JCoul| = 700–800 cm^−1^). In contrast, PDI dimers with flexible linking (Th-PDIs) exhibited greatly weakened EC (|JCoul| = 100–700 cm^−1^) than corresponding PDI dimers with fused linking. Compared to Th-FPDIs, inter-PDI geometry with non-fused linking can derive from the optimal geometry pre-defined by Th-based cores, leading to weakened EC. We further defined (JCoul^S1^ − JCoul^S0^)/JCoul^S0^ as an indicator for evaluating JCoul changing between S_0_ and S_1_ geometry.
As shown in Figure 2b, PDI dimers with fused-linking exhibited minimized JCoul changing (<10%), which is highly different from the Th-PDIs with up to 110% changing due to substantial S_1_/S_0_ ES-SR quantified by total reorganization energy of S_1_→S_0_ transition (Γ_S1→S0_). In the sense of rational molecular designing, Th-FPDIs with nearly identical JCoul on S_0_ and S_1_ states are highly appreciated as S_1_/S_0_ ES-SR might be less considered. Regarding PDI dimers with Ph-based cores, it can be seen that meta-fusing resulted in stronger inter-PDI EC (|JCoul| up to 900 cm^−1^) than corresponding Ph-pFPDIs (|JCoul| = 10–100 cm^−1^, Figure 2c). Meanwhile, Ph-mFPDIs exhibited larger S_1_/S_0_ ES-SR (Γ_S1→S0_) than corresponding para-fused dimers, leading to S_1_/S_0_ changing of JCoul up to 100% (Figure 2d). Thus, para-fused linking for Ph-based PDI dimers leads to weak inter-PDI EC, although it might be favored in the sense of rational molecular designing.
As described in Equation (1), angular factor (κ) and inter-PDI distance (R) play a central role in determining inter-PDI JCoul, for which we plotted a contour map of κ/R^3^ as a function of κ and R. As can be seen in Figure 2e, Th-FPDI, Th-PDI, Ph-mFPDI, and Ph-pFPDI dimers are located in different areas of the κ–R map, leading to their own feature on absolute values and S_1_/S_0_ changing of inter-PDI JCoul. The narrow κ (−0.6 to −0.9) and R (7–9 Å) distribution of Th-FPDI dimers resulted to considerable inter-PDI JCoul. Meanwhile, non-fused linking and substantial S_1_/S_0_ ES-SR of Th-PDI dimers lead to κ ranging from −0.2 to −1.2 with slightly enlarged inter-PDI R (9.5–11.5 Å), corresponding to greatly reduced JCoul than Th-FPDI dimers.
On the other hand, the weakened |JCoul| of Ph-pFPDIs (R = 12–13 Å) in comparison with Ph-mFPDI dimers (R = 6–7 Å) can be attributed to the greatly enlarged inter-PDI R rather than changed κ. Meanwhile, it can be seen that each Ph-pFPDI dimer exhibited nearly identical κ and R on S_0_ and S_1_ states, indicating greatly suppressed S_1_/S_0_ ES-SR in comparison to Ph-mFPDI dimers. Our calculation on JCoul of PDI dimers indicated that S_1_/S_0_ ES-SR might be a critical factor to be considered for rational molecular designing of multi-PDI systems, for which we further analyzed the origin of S_1_/S_0_ ES-SR in the sense of vibronic coupling effect in S_1_→S_0_ transition.
2.3. Vibrational Analysis
As discussed above, S_1_/S_0_ ES-SR is a critical factor for rational molecular designing of multi-PDI systems. In our recent works [31,70,71], we demonstrated that specific S_1_/S_0_ ES-SR motions can be associated with promoting vibrational modes, which can engage in S_1_→S_0_ electronic transition with high Huang-Rhys factor (Sk) and reorganization energy contribution (λ_k_). For a multi-chromophore system, inter-chromophore JCoul might also be sensitive to specific vibrational modes corresponding to collective motions that can vary inter-chromophore geometry [31,65]. To learn details on S_1_/S_0_ ES-SR of PDI dimers, we performed vibrational analysis on involved Th- and Ph-based cores, as well as corresponding PDI dimers. The Huang-Rhys factor (Sk) and reorganization energy contribution (λ_k_) of each vibrational mode (k) engaged in S_1_→S_0_ electronic transition were calculated as Sk = (2ℏ)^−1^ω_k_ΔQk^2^ and λ_k_ = 2^−1^ω_k_^2^ΔQk^2^ by using the MOMAP program [72,73,74,75] based on electronic structure calculation. The mode displacement (ΔQk) can be calculated with displacement components (ΔD_i_) along internal coordinate i as ΔQk = ∑iζ_kiΔ*D_i*.
By summing the contribution of each mode, i.e., Γ_S1→S0_ = Σ_k_λ_k_, the total reorganization energy of the S_1_→S_0_ transition (Γ_S1→S0_) can be used for generalizing S_1_/S_0_ ES-SR of PDI dimers. As listed in Table 3, all Th- and Ph-based cores exhibited considerable Γ_S1→S0_ (up to 7000 cm^−1^). Meanwhile, the calculated Γ_S1→S0_ of corresponding PDI dimers was observed to be greatly reduced to 1000–2000 cm^−1^, indicating the suppressed λ_k_ of promoting modes associated with collective motion of PDI units, which is consistent with less pronounced S_1_/S_0_ ES-SR observed by RMSD_S1/S0_. Since the collective motion of PDI units is largely correlated with low-frequency modes of Th- and Ph-cores, we could resolve S_1_/S_0_ ES-SR affecting inter-PDI geometry by extracting corresponding modes of Th- and Ph-cores. The calculated results of vibrational analysis for Th- and Ph-cores can be seen in Figure 3 and Figure 4, together with corresponding PDI dimers.
As shown in Figure 3, Γ_S1→S0_ of Th_4_ core was dominated by a promoting mode (Sk = 73.7 cm^−1^, λ_k_ = 3020 cm^−1^) at ω_k_ = 14.1 cm^−1^ (noted as mode 1, blue colored), corresponding to planarized motion on S_1_ state (see Figure S4). In the S_1_→S_0_ transition of Th_3_, Me_4_Th_3_, and Me_4_Si_2_Th_3_ cores, mode 1 might be suppressed due to the structural tension. The Me_4_Th_3_ and Me_4_Si_2_Th_3_ cores exhibited an extra promoting mode with considerable λ_k_ contribution at ω_k_ = 100–140 cm^−1^ (noted as mode 2, red colored), corresponding to twisting motion of Th units (see Figure S4). The vibrational analysis on PDI dimers indicated that λ_k_ contribution of modes 1 and 2 was greatly reduced in the S_1_→S_0_ transition of all Th-based PDI dimers, implying suppressed S_1_/S_0_ ES-SR associated with modes 1 and 2, which ensures the inter-PDI geometry pre-defined by linking cores.
Intriguingly, we observed a new promoting mode located in ω_k_ < 10 cm^−1^ region (noted as mode 3, green colored) with considerable Sk and contribution to Γ_S1→S0_ of Th-PDI dimers. As can be seen in Figure S4, mode 3 of Th-PDI dimers exhibited the largest displacement toward the opposite direction in the ending sides of PDI units, as well as the minimized displacement in spatial center of PDI units, corresponding to a rotational motion of PDI units along the linking bond between PDI and Th cores. Such a rotational motion can be greatly limited by fused linking in Th-FPDIs, for which inter-PDI geometry can be pre-defined by Th cores. However, with single bond linking, the presence of mode 3 can lead to considerable S_1_/S_0_ ES-SR of Th-PDI dimers, resulting in greatly relaxed inter-PDI geometry with undermined |JCoul|.
We subsequently performed vibrational analysis on the S_1_→S_0_ transition of the Ph-based cores and corresponding PDI dimers. As shown in Figure 4, Ph-based cores exhibited very similar promoting modes of Th-based cores discussed above, i.e., mode 1 (ω_k_ = 48.1 cm^−1^) for Ph_4_ core and mode 2 (ω_k_ = 100–120 cm^−1^) for Th_3_, Me_4_Th_3_, and Me_4_Si_2_Th_3_ cores, corresponding to planarized and Ph twisting motions (see Figure S5), respectively. With the fused linking, Ph-mFPDI and Ph-pFPDI dimers exhibited an ignorable contribution of low-frequency modes to Γ_S1→S0_, indicating minimized S_1_/S_0_ ES-SR, which can ensure inter-PDI geometry pre-defined by linking cores. Please note that vibrational modes in the high-frequency regime (>500 cm^−1^) can also contribute to Γ_S1→S0_ as well. However, high-frequency modes usually correspond to localized motions without affecting inter-PDI geometry for JCoul, which can be ignored in the discussion of the present work.
2.4. Further Discussion
As mentioned above, inter-PDI EC plays a key role in determining the excited-state relaxation path of multi-chromophore PDI model systems, in which JCoul is highly sensitive to inter-PDI geometry, i.e., angular factor κ and inter-PDI distance R. As shown in Figure 5a, we plotted a contour map of κ as a function of inter-PDI α and θ angles. The κ of PDI dimers investigated in this work ranges from −1.0 to 0.5, leading to |JCoul| tuning between 0 and 1000 cm^−1^, which is a considerable tuning range and may benefit from our strategy of molecular designing. By using rigid cores and fused covalent linking, the inter-PDI geometry of PDI dimers can be pre-defined by the topological characteristics of linking cores. As a result, collaborative modification of both inter-PDI α and θ angles can be achieved, leading to effective tuning of JCoul.
As shown in Figure 5d, collaborative changing of inter-PDI α (0°–90°) and θ (90°–0°) corresponds to a broad range of κ (1 to −3), which might lead to highly effective tuning of inter-PDI JCoul. For instance, inter-PDI α and θ in our designed PDI dimers can be changed in a relatively small range, i.e., 45°–75° and 75°–45°, respectively. However, collaborative changing of inter-PDI α and θ leads to a broad κ range (0.5 to −1.0), which is broader than the theoretically maximized tuning range by α (κ = 1 to 0) and θ (κ = 1 to −0.2) individually. As a result, a broad κ range by collaborative changing of inter-PDI α and θ can enable highly effective tuning of inter-PDI JCoul.
Last but not least, the plausible excited-state relaxation path of investigated PDI dimers can be discussed with the calculated JCoul and JCT. As shown in Figure 6, Th-FPDI dimers mostly exhibited large |JCoul|, large |JCT|, and small |JCoul + JCT|, which is consistent with the condition for plausible electronic state mixing described by Hariharan, Wasielewski, and co-workers [2,3,4,76]. In this type of PDI dimer, Th_4_-FPDI has been experimentally demonstrated to undergo SF to generate ^1^(TT) species by our ultrafast spectroscopic investigation [65], which can be regarded as the dephasing product of the mixed state. Furthermore, Ph-pFPDI dimers and most of Th-PDI dimers exhibited small |JCoul|, small |JCT|, and long inter-PDI distance, which are highly plausible to relax through incoherent SB-CS in a highly polar medium. For Ph_4_-mFPDI with huge |JCoul| (~900 cm^−1^) and close inter-PDI stacking, the excimer-like state can be expected through an incoherent relaxation.
3. Calculational Methods
All electronic structure calculations of designed cores and PDI-based dimers were performed using the Gaussian 16 software package [77]. The geometric structure of the investigated emitters was optimized on both ground (S_0_) and singlet (S_1_) excited states at B3LYP/6-311G** level, while no imaginary frequency was found by frequency analysis. The DFT and TDDFT optimized geometries of PDI-based dimers with root of the mean of squared displacement were analyzed and rendered by using the VMD 1.9.3 program [78]. The molecular orbitals analysis of the corresponding excited states was performed by using the Multiwfn program [68,69]. The Huang-Rhys (HR) factor (Sk) and reorganization energy contribution (λ_k_) of each vibrational mode were calculated by using MOMAP 2021A on the basis of frequency analysis of corresponding states [72,73,74,75]. We further estimated the reorganization energy contribution (λ_k_) of each vibrational mode between the S_1_ and S_0_ states.
4. Conclusions
To summarize, we performed molecular designing of a series of PDI covalent dimers with rigid linking cores containing thiophene (Th) or phenyl (Ph) fragments and investigated the inter-PDI electronic coupling (JCoul and JCT). The vibrational analysis on S_1_→S_0_ transition indicated that the S_1_/S_0_ ES-SR of rigid linking cores can be further suppressed by linking with PDI units, leading to comparable |JCoul| on S_0_ and S_1_ states, for which inter-PDI geometry can be pre-defined by rigid linking cores. Benefiting from the saddle-shaped feature of involved linking cores, our strategy enabled collaborative tuning of inter-PDI dihedral (α) and slipping (θ) angles, which can lead to effective tuning of inter-PDI |JCoul| = 0–1000 cm^−1^. Our work provides a new molecular designing strategy for fine-tuning of inter-chromophore EC for organic chromophores like PDI without disturbing S_1_/S_0_ ES-SR, which might be useful for future applications in organic optoelectronics.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Le A.K. Bender J.A. Arias D.H. Cotton D.E. Johnson J.C. Roberts S.T. Singlet Fission Involves an Interplay between Energetic Driving Force and Electronic Coupling in Perylenediimide Films J. Am. Chem. Soc.201814081482610.1021/jacs.7b 1188829240411 · doi ↗ · pubmed ↗
- 2Young R.M. Wasielewski M.R. Mixed Electronic States in Molecular Dimers: Connecting Singlet Fission, Excimer Formation, and Symmetry-Breaking Charge Transfer Acc. Chem. Res.2020531957196810.1021/acs.accounts.0c 0039732786248 · doi ↗ · pubmed ↗
- 3Lin C. Kim T. Schultz J.D. Young R.M. Wasielewski M.R. Accelerating Symmetry-Breaking Charge Separation in a Perylenediimide Trimer through a Vibronically Coherent Dimer Intermediate Nat. Chem.20221478679310.1038/s 41557-022-00927-y 35469005 · doi ↗ · pubmed ↗
- 4Lijina M.P. Benny A. Sebastian E. Hariharan M. Keeping the Chromophores Crossed: Evidence for Null Exciton Splitting Chem. Soc. Rev.2023526664667910.1039/D 3CS 00176 H 37606527 · doi ↗ · pubmed ↗
- 5Miyamoto H. Okada K. Tada K. Kishi R. Kitagawa Y. Theoretical Study on Singlet Fission Dynamics and Triplet Migration Process in Symmetric Heterotrimer Models Molecules 202429544910.3390/molecules 2922544939598837 PMC 11597243 · doi ↗ · pubmed ↗
- 6Gao F. Xu Y. Hou L. Liu D. Yang Z. Ding Z. Meng P. Liu H. Zhang J. Zhao Z. In-Solution Intramolecular through-Space Conjugations of Sterically Constrained Tetranaphthylethane CCS Chem.202511210.31635/ccschem.025.202405283 · doi ↗
- 7Martin I.J. Masese F.K. Shih K.-C. Nieh M.-P. Kasi R.M. Nanoscale “Chessboard” Pattern Lamellae in a Supramolecular Perylene-Diimide Polydiacetylene System Molecules 202530120710.3390/molecules 3006120740141984 PMC 11946615 · doi ↗ · pubmed ↗
- 8Wu Y. Zhou J. Phelan B.T. Mauck C.M. Stoddart J.F. Young R.M. Wasielewski M.R. Probing Distance Dependent Charge-Transfer Character in Excimers of Extended Viologen Cyclophanes Using Femtosecond Vibrational Spectroscopy J. Am. Chem. Soc.2017139142651427610.1021/jacs.7b 0827528880547 · doi ↗ · pubmed ↗