Retinal Changes in Early-Onset cblC Methylmalonic Acidemia Identified Through Expanded Newborn Screening: Highlights from a Case Study and Literature Review
Paola Michieletto, Francesco Baldo, Maurizio Madonia, Luisa Zupin, Stefano Pensiero, Maria Teresa Bonati

TL;DR
This case study shows that retinal degeneration can occur in newborns with cblC methylmalonic acidemia, even with early treatment, highlighting the need for immediate therapy and close eye monitoring.
Contribution
The study emphasizes the importance of early ophthalmological monitoring in cblC patients to detect retinal changes before visual symptoms appear.
Findings
Retinal degeneration was detected at 3 months in a newborn with cblC despite early treatment.
Bull’s eye maculopathy and reduced ERG amplitudes were observed at 7 months.
Visual field testing showed no abnormalities despite retinal changes.
Abstract
Background: Methylmalonic acidemia combined with homocystinuria (cblC) can lead to infantile maculopathy. Although significant visual deterioration is commonly reported in early-onset cblC, we found poor awareness regarding formal assessments of ocular complications, especially in newborns, and of how these complications relate to the timing of therapy initiation. In this work, we present our experience and perform a literature review. Methods: We performed sequential fundus examinations, optical coherence tomography (OCT) and full-field electroretinography (ERG) under sedation following detection of signs of retinal degeneration. We also assessed visual fields using kinetic attraction perimetry. Results: We report a newborn who was referred on the eighth day of life, following a diagnosis of cblC through newborn screening (NBS), and who began treatment that same day. Close monitoring…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
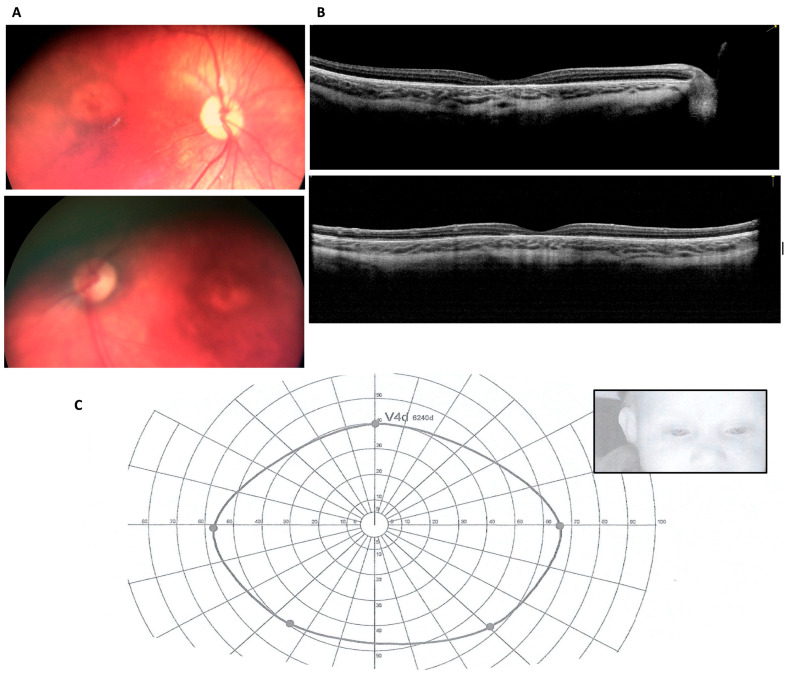 Figure 1
Figure 1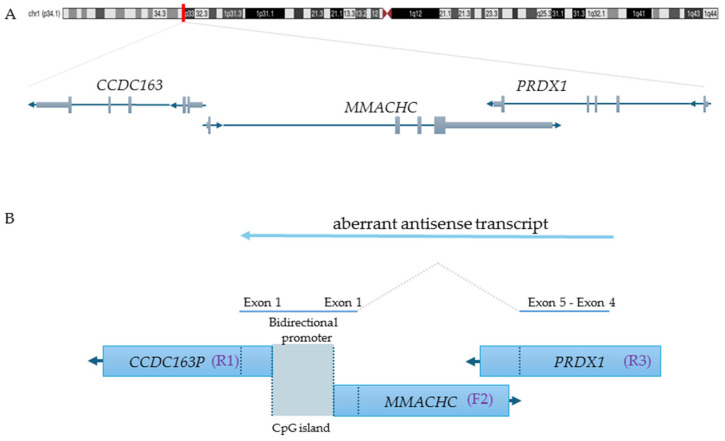 Figure 2
Figure 2- —Italian Ministry of Health
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMetabolism and Genetic Disorders · Folate and B Vitamins Research · Neonatal Health and Biochemistry
1. Introduction
Combined methylmalonic acidemia and homocystinuria (OMIM #277400, MAHCC), also known as cblC, is the most common inborn error of intracellular cobalamin (vitamin B_12_) metabolism, with a prevalence of 1:37,000 to 1:100,000. In Italy, cblC was introduced in newborn screening (NBS) between 2016 and 2017 [1]. NBS allows detection of patients, often before the onset of symptoms, and early treatment.
CblC results from a deficiency of the MMACHC chaperone, which impairs the conversion of vitamin B_12_ into its two active forms, adenosylcobalamin and methylcobalamin. These serve as cofactors for two enzymes, methylmalonyl-CoA mutase (which converts methylmalonyl-CoA into succinyl-CoA in mitochondria) and methionine synthase (which catalyzes in the cytosol the conversion of homocysteine into methionine), whose dysfunction leads to elevated levels of methylmalonic acid (MMA) and homocysteine (Hcy) in plasma and urine, along with normal or reduced plasma methionine (Met) levels [2].
The disorder is inherited in an autosomal recessive manner. It can be either monogenic, with bi-allelic pathogenic variants in MMACHC, or digenic, with one of the two pathogenic variants located in the adjacent PRDX1 gene, encoding peroxiredoxin 1, involved in cell defense against oxidative stress with effect on cell growth, differentiation and apoptosis. Phenotypically, digenic cblC is nearly indistinguishable from the monogenic form, as pathogenic variants in PRDX1—of splicing type—lead to aberrant PRDX1 antisense activity, causing cys-hypermethylation of the MMACHC promoter and subsequent transcriptional silencing [3]. Since this mutational event results from an epigenetic modification at the MMACHC locus, digenic cblC is called ‘epi-cblC’.
CblC can present with a broad spectrum of clinical features and age of presentation. Patients with early-onset disease (<12 months) may have feeding difficulties, failure to thrive, developmental delay, hypotonia, seizures, anemia, thrombocytopenia, neutropenia, and microangiopathy. Late-onset disease presents after the first year of life with neurologic dysfunction such as cognitive decline, psychiatric disturbances, and subacute degeneration of the spinal cord. In the recently recognized prenatal form, the symptoms may appear during pregnancy or shortly after birth and include fetal growth retardation, and neurological and developmental issues.
Patients with prenatal and early-onset cblC may have significant ocular features, whereas visual loss and ocular complications are rare and mild in the late-onset form. The typical pattern of retinal degeneration is characterized by bilateral macular atrophy with a bull’s-eye pattern, i.e., a hypopigmented perimacular zone surrounded by a hyperpigmented ring. These marks can be seen on fundoscopy and optical coherence tomography (OCT). In addition to maculopathy, the ophthalmic phenotype includes pigmentary retinopathy and, less frequently, optic atrophy. Ocular findings are strabismus, nystagmus and/or visual inattention [2,4].
Therapy cannot entirely prevent ocular symptoms, even in patients identified by NBS or prenatal diagnosis and biochemical abnormalities aggressively treated early/in utero. Although the natural history of cblC is well known—with visual deterioration and blindness being common outcomes in early-onset patients—there is still limited awareness regarding the formal assessment of ocular complications, particularly timing and the types of ophthalmological evaluations required [4]. Typically, an ophthalmological examination is only recommended at diagnosis [2].
We report on the onset and progression of ocular disease measured by ERG and OCT in a seemingly healthy little girl who underwent treatment after biochemical diagnosis of a cobalamin defect at NBS. Prompted by the patient’s history and a review of cases with early-onset cblC and ocular involvement, we realized that certain ophthalmological features, such as nystagmus, should be further categorized to assess the impact of early therapeutic intervention. We recommend that neonates with a positive NBS for MMA and confirmed hyperhomocysteinemia begin treatment without delay and be promptly referred to a pediatric ophthalmologist experienced in inherited metabolic disorders.
2. Materials and Methods
2.1. Patient
Based on NBS results, the newborn patient was suspected of having a cobalamin C/D defect and subsequently referred to the Neonatology Department to the Pediatric Clinic of Maternal and Child Health IRCCS Burlo Garofolo for further investigation and treatment. Visual and ophthalmologic evaluations were carried out at the hospital’s Ophthalmology Department.
2.2. Newborn Screening and Confirmatory Testing
NBS samples of dried blood spots (DBSs) were collected between 48 and 72 h, according to the recommended time frame. Blood levels of propionylcarnitine (C3), MMA and Hcy were measured by tandem mass spectrometry at the University Hospital of Padova. Second DBSs were requested within 24 h upon abnormal first-screening results [1]; in this second test, the second-tier marker included Met. Plasma Hcy, acylcarnitines, MMA and Met were measured to confirm the diagnosis further. Plasma vitamin B_12_ levels were determined to rule out a maternal deficiency.
2.3. Molecular Analysis
Whole exome sequencing (WES) of the proband and parents (trio WES) was performed at the Medical Genetics Laboratory of IRCCS Burlo Garofolo as described in Feresin et al. [5].
2.4. Visual and Ophthalmologic Testing
We performed optical coherence tomography (OCT) and full-field electroretinography (ERG) under sedation, and measured the extension of the binocular visual field by means of ‘Attraction perimetry’, an advanced evolution of the Metrovision (MonCv3) system, designed to detect and quantify early campimetric alterations in glaucoma [6]. The binocular attraction visual field was performed using Goldmann V4 stimuli and a background brightness of 10 candles/m^2^.
2.5. Literature Review
We reviewed pathogenic variants with the support of online databases, including ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, accessed on 18 March 2025), and HGMD^®^ Professional (https://digitalinsights.qiagen.com/) (accessed on 18 March 2025).
Moreover, we extracted data from case reports and case series of patients affected by infantile cblC to compare retinal disease features, treatment initiation and outcome with those of our patient. PubMed search terms included ‘maculopathy’, ‘ocular’, ‘retinal’, and ‘retinopathy’. Each was combined with the search term ‘cblc type methylmalonic acidemia’ (accessed on 6 February 2025). We excluded from the review investigations focusing on late-onset cblC, as well as animal and in vitro studies.
3. Results
3.1. Clinical Report
The patient was the first offspring of healthy unrelated Italian parents of Caucasian ethnicity and was born at term by vaginal delivery after normal pregnancy. The mother’s first pregnancy resulted in termination due to gastroschisis; the second pregnancy resulted in spontaneous termination in the second trimester. Family history was otherwise not contributory. Her birth weight was 2790 g (10–25th centile), birth length 49 cm (25–50th centile), and occipitofrontal circumference (OFC) 33 cm (10–25th centile).
On the eighth day after birth, she was referred for study following biochemical diagnosis of cblC/cblD defect by NBS (Table 1). The patient exhibited slight drowsiness and poor sucking, resulting in mild dehydration as suggested by dry skin (at the turgor test, it took the skin 3 s to return to its original position) and mucous membranes, and decreased diuresis collected in the anamnesis. At the neurological examination she was found to have moderate axial and peripheral hypotonia. She was started on intramuscular hydroxocobalamin (OHCbl) 1 mg a day (1 mg/mL solution), after drawing for blood tests (Table 1) and having ruled out maternal vitamin B_12_ deficiency. Biochemical tests also included blood count, capillary blood gas analysis and urine stick, which were found average, and ammonium dosage, which was 113 µg/dL (nv 27–90). The baby promptly recovered, and starting the following day, betaine 100 mg was administered three times daily, along with folic acid 5 mg twice a week. Otoemissions, automated auditory brainstem responses (AABR), electrocardiogram, echocardiogram, and transfontanellar ultrasound performed at 9 and 13 days of age, were all normal.
She underwent fundus examination on the ninth day which was normal. At the age of 3 months, she did not exhibit any pupillary defects nor strabismus or alterations in ocular motility. She had a good fixation and normal pursuit. Cycloplegic refraction in dioptres was slightly astigmatic (+2.00 cyl/70° in the RE and +1.50 cyl/100 TABO in the LE). Fundus examination revealed a mild pigmentary perifoveal dystrophy with a standard optic disc.
At 5 months, the fundus examination showed central macular atrophy with only slightly pale optic disc. At 7 months the evaluation was performed under sedation: a bilateral bull’s eye maculopathy was present (Figure 1A); OCT images (Optovue iVue, Freemont, CA, USA) (Figure 1B) showed a very thin fovea with an Outer Plexiform Layer (OPL) that became even thinner when moving toward the fovea, until it disappeared in the atrophic area that involved only the external layers. The full-field ERG showed an amplitude reduction of both scotopic and photopic components. We performed a binocular Kinetic attraction visual field (Figure 1C) some days later, resulting in a standard (bilateral temporal extension of 60°).
At the last follow-up, at the age of 10 months, her psychomotor development was within normal limits. As for growth parameters, weight was 7.4 kg (3rd centile), length 70.5 cm (27th centile), and OFC 43 cm (9th centile).
3.2. Biochemical Features
As shown in Table 1, with therapy Hcy values gradually decreased to slightly above the normal range; C3 and MMA values averaged and Met increased. These data document the achievement of a good biochemical control. OHCbl was administered by a subcutaneous catheter device [7] for 4 months, with success, to avoid multiple punctures. We removed the subcutaneous injection port due to the parents’ concern about infections at the injection site, and intramuscular therapy was resumed.
3.3. Genetic Findings
The patient was found to be compound heterozygous for the pathogenic variants MMACHC (NM_015506.3): c.331C>T, p.(Arg111*) [8,9] in exon 3 and PRDX1 (NM_002574.4): c.515-1G>T [3,9], inherited from the father and the mother, respectively. Genetic findings fit with the patient’s biochemical and clinical features.
The MMACHC (NM_015506.3): c.331C>T is a nonsense substitution that truncates the protein at codon 111, which is 172 amino acids from the end of the protein. The nonsense variant was classified as pathogenic according to the ACGS/ACMG-AMP criteria PVS1, PM2_moderate, PP3 and PP5_supporting (https://wintervar.wglab.org/, accessed on 30 December 2024).
The PRDX1 (NM_002574.4): c.515-1G>T is a splicing variant that activates a cryptic acceptor site in intron 5, producing the skipping of the last exon (6) and the loss of transcription termination signal of PRDX1 [3].
3.4. Digenic Methylmalonic Acidemia and Homocystinuria, cblC Type (epi-cblC)
In epi-cblC, the subjects are compound heterozygotes for a pathogenic variant in an allele of the disease-causing gene (MMACHC) and a secondary epimutation in the promoter of the other allele of MMACHC. The epimutation results from a splicing defect in the PRDX1 gene, caused either by c.515-1G>T, as seen in our patient, or by c.515-2A>T, which are located in the canonical splice acceptor site in intron 5.
MMACHC belongs to a gene trio on the 1p34.1 chromosome region, in which it is a sense gene flanked by CCDC163P (a pseudogene) and PRDX1 in the opposite orientation (trio reverse(R1)/forward(F2)/reverse(R3)) (Figure 2A).
Each PRDX1 splicing variant extends the antisense transcription across PRDX1 exons 4 and 5, part of MMACHC exon 1, the bidirectional promoter of the adjacent MMACHC/CCDC163P genes, and part of the first exon of CCDC163P, as a result of the activation of a cryptic antisense splicing site within the MMACHC exon 1 (Figure 2B). This aberrant transcription may cause RNA polymerase collision and the formation of triplexes, leading to the methylation of the gene promoter and the consequent silencing of the gene expression [3,9].
Although epi-cblC is characterized by compound epigenetic-genetic heterozygosity with haploinsufficiency of MMACHC, it is classified as digenic. This arises from the inability to exclude the possibility that PRDX1 splicing variants may have deleterious effect on peroxiredoxin 1, thereby potentially affecting the disease course.
As a unique example among human diseases, in cblC, the MMACHC epimutation is inherited in cis with the pathogenic splicing variant of PRDX1 [3].
3.5. Literature Mining
The [MMACHC (NM_015506.3): c.331C>T, p.(Arg111*); PRDX1 (NM_002574.4): c.515-1G>T] genotype has been described in a male from Northern Italy –10 years at last follow-up– with early-onset disease, who started treatment with enteral feeding, red blood cells transfusions, OHCbl, L-carnitine, betaine and folates from 2 months, when he was diagnosed after a clinical/biochemical assessment [9]. The boy was born before the implementation of NBS, so a precocious diagnosis, prior to symptom onset, was not possible. Briefly, his clinical history began on the fourth day with sucking difficulties and recurrent vomiting, then severe weight loss on the twenty-first day. He underwent hospitalization because of severe failure to thrive and vomiting with subsequent drowsiness, hypotonia, sepsis, respiratory distress and acute neurological crisis between 42 and 104 days after birth. A brain MRI at age 2 months showed frontotemporal atrophy with hygromas. He exhibited nystagmus, macular hypoplasia, and exotropia/exophoria at 4 months, 6 and 7 years, respectively. At the age of 3 years, he was diagnosed with a moderate intellectual disability [9].
In cblC a genotype-phenotype correlation has not been identified. However, the genotype homozygous for the common c.271dupA allele is characterized by early-onset phenotype and early childhood-onset maculopathy. Moreover, the presence of homozygous or compound heterozygous frameshift and non-sense genetic variants are often associated with a more severe phenotype.
Through the PubMed search, we retrieved 15 manuscripts [4,8,10,11,12,13,14,15,16,17,18,19,20,21]. Some of them were excluded from the discussion because they dealt with late-onset cblC [12,18] and animal [19] or in vitro [17] disease models. In particular, for the study, the manuscripts by Weisfeld-Adams et al. [4] and Ku et al. [11] were especially valuable, since they reported when patients were started on therapy and included those treated prenatally.
Indeed, prenatal molecular diagnosis—through target sequencing of pathogenic variants in both copies of MMACHC or one copy of both MMACHC and PRDX1 using DNA extracted from chorionic villi or amniocytes—may be proposed where an older sibling is affected. Additionally, intramuscular hydroxocobalamin may be administered to the mother during the third trimester of pregnancy if pathogenic variants are identified in prenatal diagnosis.
Alternatively, couples may be offered preimplantation genetic diagnosis through assisted reproductive technology.
Table 2 displays the patients selected for comparison and discussion: we included early-onset cblC patients who began therapy within the first month of life, also considering in this category those reported as having initiated treatment ‘immediately after diagnosis’.
4. Discussion
In newborns or infants, ocular involvement, especially retinal degeneration, is frequent among the earliest detectable signs of early-onset cblC type methylmalonic acidemia with homocystinuria. As in the infant here described, macular atrophy may be the first sign observed, even without evidence of visual deprivation, such as nystagmus. Therefore, in cblC macular atrophy may be overlooked if not actively sought, for instance through OCT, since it can detect even the most subtle and asymptomatic changes of maculopathy.
Furthermore, by exhibiting macular atrophy as early as 5 months of age, despite prompt initiation of therapy and adequate biochemical control, this clinical report provides further evidence of the very early onset of retinal disease in epi-cblC-type methylmalonic acidemia [9].
Conversely, macular atrophy may be overlooked when systemic issues, such as hypotonia and poor feeding, may take precedence over a detailed eye examination.
Some authors have hypothesized that maculopathy in infantile cblC likely develops before birth.
The exact mechanism of the specific eye involvement in cblC remains unclear. Since a critical phase of foveal development occurs postnatally, extending through infancy into early childhood, the fovea may be particularly vulnerable to the toxic accumulation of homocysteine and methylmalonic acid during this window, potentially explaining the rapid progression of macular alteration observed in affected patients [4,14]. Additionally, ocular tissues are particularly vulnerable to metabolic disturbance due to their high energy requirement. A deficiency in the MMACHC protein impairs the intracellular processing of vitamin B_12_ into adenosylcobalamin, a cofactor for the methylmalonyl-CoA mutase. This mitochondrial enzyme is a component of the propionate catabolic pathway, which metabolizes branched-chain amino acids, odd-chain fatty acids and the side chain of cholesterol. Methylmalonyl-CoA mutase catalyzes the conversion of methylmalonyl-CoA into succinyl-CoA that enters the tricarboxylic acid cycle, serving as an anapleurotic substrate [26].
In the outer retina, photoreceptors, retinal pigment epithelium and Müller cells possess a high density of mitochondria to support their metabolic activity. Furthermore, retinal ganglion cells have unmyelinated axons until they pass through the lamina cibrosa. In this unmyelinated region, the generation and propagation of action potentials demand substantial energy supplied by a high mitochondrial content [27]. Moreover, photo-oxidative stress makes ocular tissues more susceptible to oxidative damage [28]. Furthermore, although according to the Human Protein Atlas MMACHC is not specifically expressed in the retina compared to other tissues [29], the Human Eye Transcriptome Atlas shows an enrichment of MMACHC expression in the choroid and retinal pigment epithelium, suggesting, for this gene, a potential role in retinal function [30].
Eye involvement may be indirect. A post-mortem histopathological study of a cblC infant revealed, at the electron microscopy level, diffuse storage of material in cytoplasmic vacuoles across various ocular tissues, along with swollen mitochondria in the corneal epithelium and degenerated mitochondria in the iris pigment epithelium [31].
Macular changes can develop as early as before the first month of age (Table 2 and [17]), often presenting as subtle macular pigmentary or atrophic changes. These macular changes may progress to severe macular lesions at a dramatically fast space, usually within the first two years of age, but as early as 6 months of age (Table 2 and [17]).
Notably, the clinical findings in patient n. 56 [4] (Table 2) closely resemble those observed in our patient.
Among the patients listed in Table 2, five out of nineteen, including the patient reported here, exhibited retinal degeneration without developing nystagmus (Table 2). Furthermore, seven out of nineteen patients had retinal degeneration and nystagmus (Table 2), while another seven exhibited nystagmus without retinal degeneration (Table 2).
In early-onset retinal dystrophies, nystagmus is the most common sign, occurring, for instance, in 76% of the cohort of 50 children studied by Suppiej et al. [32], and it typically manifests within the first 6 months of life, as occurred in 34 children of the 40 who exhibited nystagmus from the same cohort [32]. The onset timing of nystagmus in retinal diseases is influenced by the severity and extent of tissue damage that occurs during the development of the fixation reflex. This reflex, which enables the eyes to reflexively orient and maintain fixation on a target, typically develops within the first three months of life. In the child here reported, the development of maculopathy after the third month of life allowed the patient to establish the fixation reflex, thereby preventing the onset of nystagmus and thus prospecting a more favourable visual outcome.
Sensory nystagmus, often of the pendular type in the early stages, can result from macular dystrophy, either congenital or occurring within the first 2–3 months of life, or congenital developmental abnormalities such as foveal or optic nerve hypoplasia.
The presence or absence of nystagmus in patients with early-onset maculopathy is related to the timing of maculopathy development relative to the fixation reflex. When maculopathy develops after the fixation reflex has already emerged, as observed in our patient, sensory nystagmus is unlikely to occur. This is consistent with sensory nystagmus patterns.
On the other hand, the development of nystagmus in the absence of maculopathy suggests the presence of different types of nystagmus in cblC patients. This aspect has not been addressed before. In fact, in the patient reported by Gerth et al. [33], nystagmus was present from birth and diagnosed as congenital motor nystagmus. Motor nystagmus, typically of the jerking type, unlike sensory nystagmus, can also be present at birth and may result from neurological abnormalities detectable at brain MRI. Additionally, congenital motor nystagmus may be idiopathic (without brain MRI abnormalities) and sometimes exhibit familiarity, usually an autosomal dominant inheritance pattern.
Therefore, it is crucial to distinguish early-onset cblC patients with nystagmus due to sensory alterations from those with motor nystagmus.
Early diagnosis and treatment, as in our patient, could influence the development of sensory nystagmus but would have no effect on motor nystagmus.
Interestingly, patient n. 6 described by Ku et al. [11] (Table 2), who was followed ophthalmologically from 8 months, showed signs of nystagmus at 8 months but developed the characteristic BEM maculopathy at 4.8 years of age. This clinical history suggests that a thorough ophthalmological follow-up is warranted for cblC patients, even after the first two years of life.
Similar to patient n. 6 [11], the fundus oculi of patient P3, as described by Schimel et al. [24] (Table 2), appeared normal upon examination at 6 months. However, evidence of photoreceptor disease was detected through ERG. Therefore, we suggest performing ERG in cblC patients even if they do not exhibit signs of maculopathy.
Retinal changes in cblC patients who underwent early treatment overlap with those diagnosed later. For example, the epi-cblC patient with the same genotype as our patient exhibited an ocular phenotype that falls between those of the first two patients listed in Table 2.
Optic atrophy may also be present in each of the three subgroups, often associated with a worse outcome. In cblC, optic atrophy may coexist with retinal disease or may present as an isolated finding [34]. In instances where optic atrophy is associated with retinal pathology, it is plausible that the atrophy results from diminished input from the retina. Specifically, in the presence of maculopathy, the optic nerve ganglion cells may undergo degeneration due to the absence of activation from their corresponding photoreceptors, particularly the cones. In our patient, it appears that the observed optic pallor is likely a secondary consequence of the underlying maculopathy. This assumption fits with the ERG findings. Conversely, it is also possible that the optic atrophy represents a primary condition, independent of retinal involvement.
Hydroxocobalamin supplementation is considered the standard therapy for cblC, with studies suggesting higher doses, especially early in treatment, may improve the biochemical and neurodevelopmental profiles. Indeed, vitamin B_12_ can cross the retinal blood barrier [35] and brain blood barrier [36], thus supporting the potential efficacy of hydroxocobalamin therapy.
However, an Italian study reported no improvement in maculopathy, with a median hydroxocobalamin dose of 0.55 mg/kg/day (range 0.30–0.90) compared to medium (0.09 mg/kg/day) and low (0.06 mg/kg/day) doses. In contrast, improvements in metabolic profile and neurocognitive outcome were found in the high dose group [37]. A Luxemburg study using higher doses (mean ± SD 6.5 ± 3.3 mg/kg/day) found better ophthalmologic and cognitive outcome in 4 infants who started treatment before 5 months of age, compared to a child who began treatment at 5 years [38]. Additionally, Venditti et al. [39] reported that five of the six patients treated with hydroxocobalamin dose intensification (ranging from 0.4 to 2.7 mg/kg/day) did not develop maculopathy or retinopathy. In contrast, within a historical cohort of 27 patients who received lower doses (≤0.3 mg/kg/day), all developed maculopathy.
The optimal dosing regimen for hydroxocobalamin in cblC has not yet been established. However, recent studies suggest that a higher dose may be more efficacious in biochemical control than a lower dose.
We suggest that neonates following a positive NBS for MMA with confirmed hyperhomocysteinemia be immediately started on cobalamin supplementation and promptly referred to a pediatric ophthalmologist with expertise in inherited metabolic diseases for early evaluation. Indeed, based on the above observations, we confirm that prompt treatment may benefit systemic disease control, although it does not prevent macular disease progression [16,20,40].
5. Conclusions
In conclusion, early-onset cobalamin C deficiency can present with significant ocular complications, such as retinal degeneration, nystagmus and optic neuropathy.
Ocular involvement may be ‘cryptic’ and can precede any systemic manifestations, making cblC a particularly insidious disease. Conversely, its onset may be delayed by years compared to the systemic onset, even if the latter occurs very early. Therefore, careful monitoring for ocular signs and symptoms is necessary throughout the course of development.
Pre-symptomatic diagnosis, primarily through newborn screening and genetic testing, allows for prompt initiation of treatment, typically involving vitamin B_12_ supplementation and other metabolic therapies, to manage the condition and mitigate its effects.
It is worth noting that genetic testing for methylmalonic acidurias must consider epi-cblC, and pathogenic variants must be searched for in MMACHC and PRDX1. Epivariation in R1/F2/R3 trios of genes is a disease pathomechanism to consider in patients exhibiting typical manifestations of a recessive disorder but bearing a single heterozygous pathogenic variant in the causal gene.
We recommend beginning therapy as soon as the biochemical defect is confirmed and prenatally in the case of a prenatal diagnosis.
In cblC there are two types of nystagmus: sensory, resulting from retinal degeneration and/or optic atrophy, and motor, which may arise from brain abnormalities or be idiopathic. Additionally, MMACHC itself may be a susceptibility gene for motor or congenital idiopathic nystagmus.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ruoppolo M. Malvagia S. Boenzi S. Carducci C. Dionisi-Vici C. Teofoli F. Burlina A. Angeloni A. Aronica T. Bordugo A. Expanded Newborn Screening in Italy Using Tandem Mass Spectrometry: Two Years of National Experience Int. J. Neonatal Screen.202284710.3390/ijns 803004735997437 PMC 9397032 · doi ↗ · pubmed ↗
- 2Huemer M. Diodato D. Schwahn B. Schiff M. Bandeira A. Benoist J.F. Burlina A. Cerone R. Couce M.L. Garcia-Cazorla A. Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cbl C, cbl D, cbl E, cbl F, cbl G, cbl J and MTHFR deficiency J. Inherit. Metab. Dis.201740214810.1007/s 10545-016-9991-427905001 PMC 5203859 · doi ↗ · pubmed ↗
- 3Guéant J.L. Chéry C. Oussalah A. Nadaf J. Coelho D. Josse T. Flayac J. Robert A. Koscinski I. Gastin I. APRDX 1 mutant allele causes a MMACHC secondary epimutation in cbl C patients Nat. Commun.2018967 Erratum in Nat. Commun. 2018, 9, 55410.1038/s 41467-017-02306-529302025 PMC 5754367 · doi ↗ · pubmed ↗
- 4Weisfeld-Adams J.D. Mc Court E.A. Diaz G.A. Oliver S.C. Ocular disease in the cobalamin C defect: A review of the literature and a suggested framework for clinical surveillance Mol. Genet. Metab.201511453754610.1016/j.ymgme.2015.01.01225742969 · doi ↗ · pubmed ↗
- 5Feresin A. Spedicati B. Zampieri S. Morgan A. Magnolato A. Tesser A. Tommasini A. Bonati M.T. Girotto G. Faletra F. Does It Run in Your Family? Inherited Truncating PSMD 12 Variants Broaden the Phenotypic Spectrum of Stankiewicz-Isidor Syndrome Am. J. Med. Genet. Part A 2024197 e 6395310.1002/ajmg.a.6395339641441 · doi ↗ · pubmed ↗
- 6Daneshvar R. Ehsaei A. Moghadas Sharif N. Pato Z. Comparison of Visual Field Measurements in Glaucomatous Eyes using Oculus and Metrovision Perimeters J. Curr. Ophthalmol.202335172210.4103/joco.joco_197_2237680285 PMC 10481978 · doi ↗ · pubmed ↗
- 7Maines E. Morandi G. Gugelmo G. Ion-Popa F. Campostrini N. Pasini A. Vincenzi M. Teofoli F. Camilot M. Bordugo A. Vitamin B 12 Administration by Subcutaneous Catheter Device in a Cobalamin A (cbl A) Patient JIMD Rep.20173529312785837310.1007/8904_2016_20PMC 5585097 · doi ↗ · pubmed ↗
- 8Gizicki R. Robert M.C. Gómez-López L. Orquin J. Decarie J.C. Mitchell G.A. Roy M.S. Ospina L.H. Long-term visual outcome of methylmalonic aciduria and homocystinuria, cobalamin C type Ophthalmology 201412138138610.1016/j.ophtha.2013.08.03424126030 · doi ↗ · pubmed ↗