Exploration of Bromodomain Proteins as Drug Targets for Niemann–Pick Type C Disease
Martina Parente, Amélie Barthelemy, Claudia Tonini, Sara Caputo, Alessandra Sacchi, Stefano Leone, Marco Segatto, Frank W. Pfrieger, Valentina Pallottini

TL;DR
This study explores bromodomain proteins as potential drug targets for Niemann–Pick Type C Disease, a fatal disorder involving cholesterol metabolism issues in lysosomes.
Contribution
The study identifies bromodomain and extra-terminal domain (BET) proteins as novel therapeutic targets for Niemann–Pick Type C Disease (NPCD).
Findings
Treatment with JQ1, a BET protein inhibitor, increased NPC1 protein levels and reduced lysosomal cholesterol accumulation.
JQ1 induced extracellular release of lysosomal components in a dose- and patient-dependent manner.
JQ1 modulated cholesterol accumulation caused by NPC1 inhibition and histone deacetylase activity.
Abstract
Defects in lysosomal cholesterol handling provoke fatal disorders presenting neurovisceral symptoms with variable onset and life spans. A prime example is Niemann–Pick type C disease (NPCD), where cholesterol export from the endosomal–lysosomal system is impaired due to variants of either NPC intracellular cholesterol transporter 1 (NPC1) or NPC intracellular cholesterol transporter 2 (NPC2). Therapeutic options for NPCD are limited to palliative care and disease-modifying drugs, and there is a need for new treatments. Here, we explored bromodomain and extra-terminal domain (BET) proteins as new drug targets for NPCD using patient-derived skin fibroblasts. Treatment with JQ1, a prototype BET protein inhibitor, raised the level of NPC1 protein, diminished lysosomal expansion and cholesterol accumulation, and induced extracellular release of lysosomal components in a dose-, time-, and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
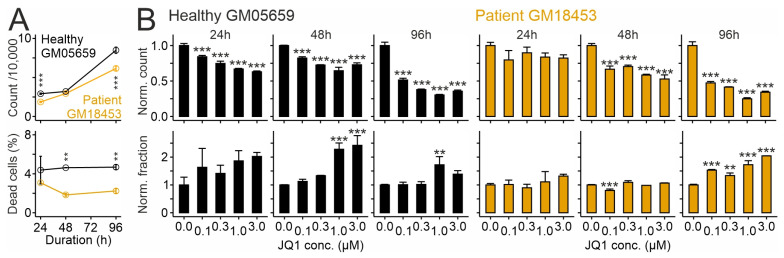 Figure 1
Figure 1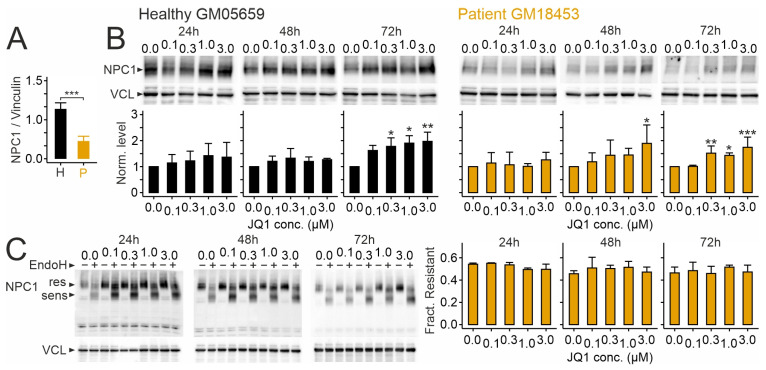 Figure 2
Figure 2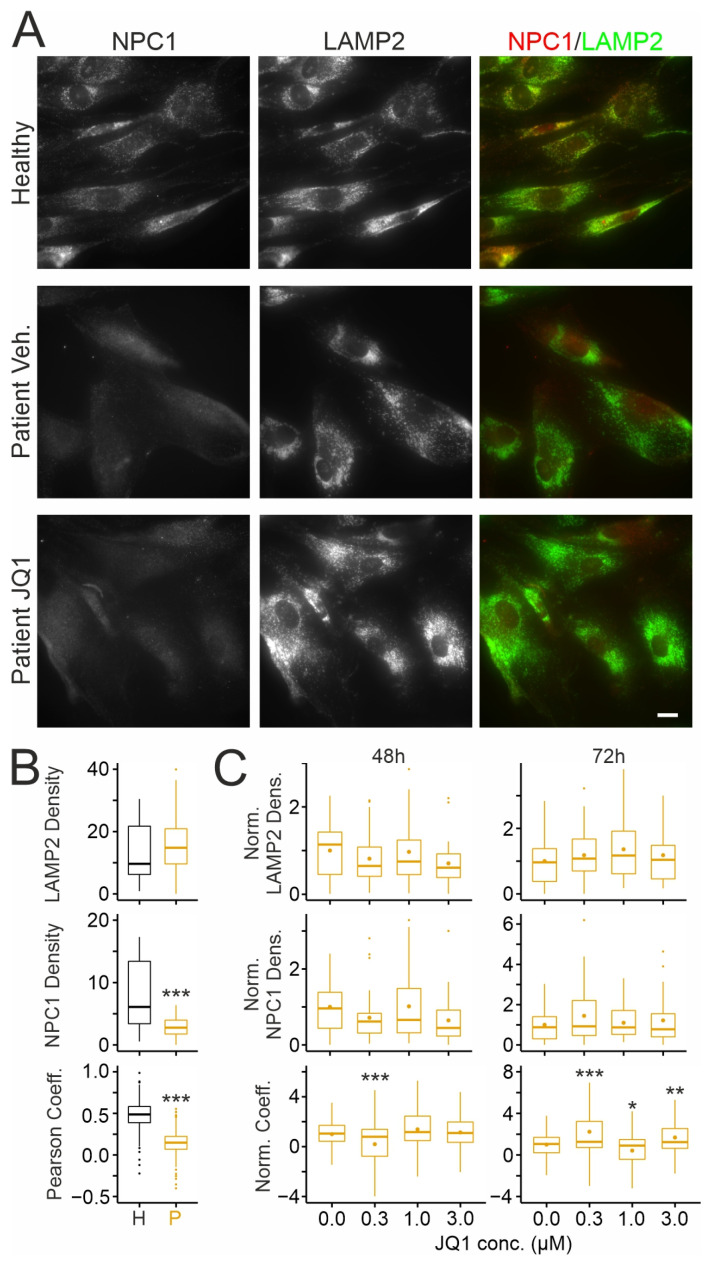 Figure 3
Figure 3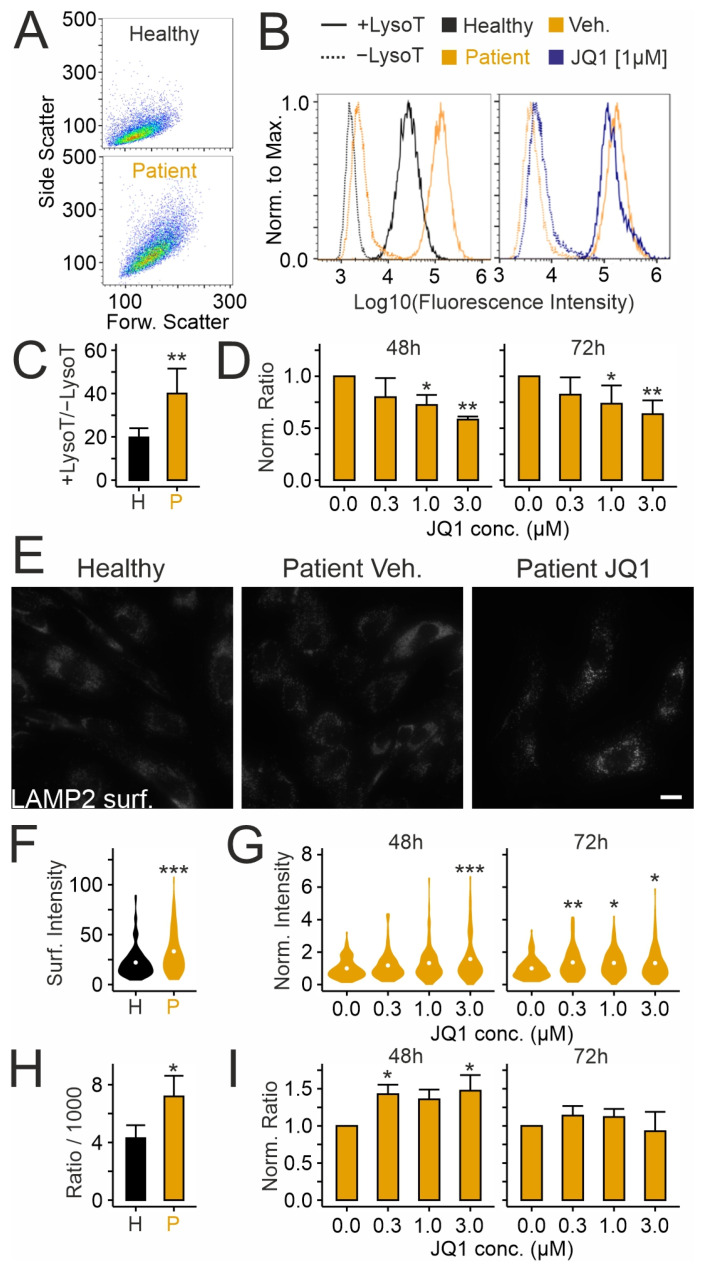 Figure 4
Figure 4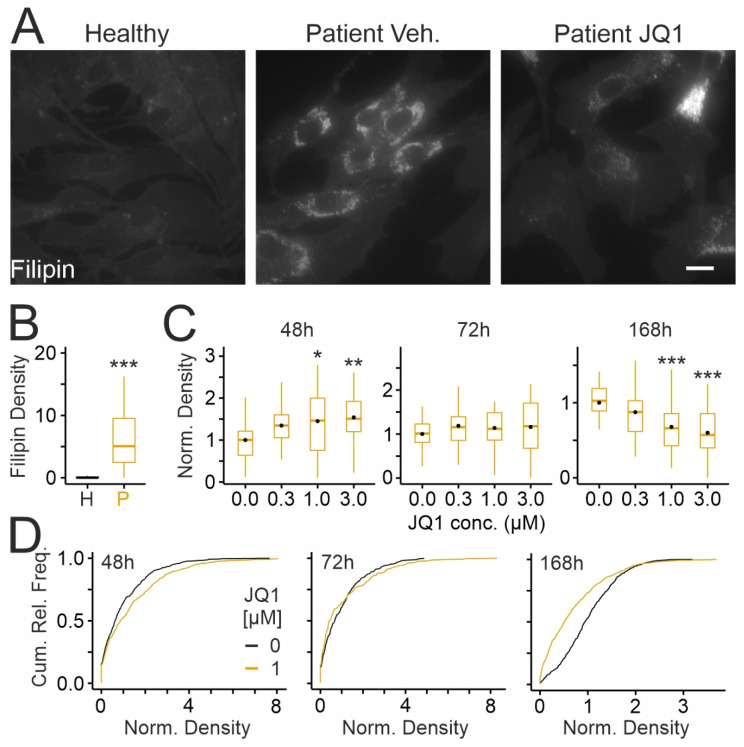 Figure 5
Figure 5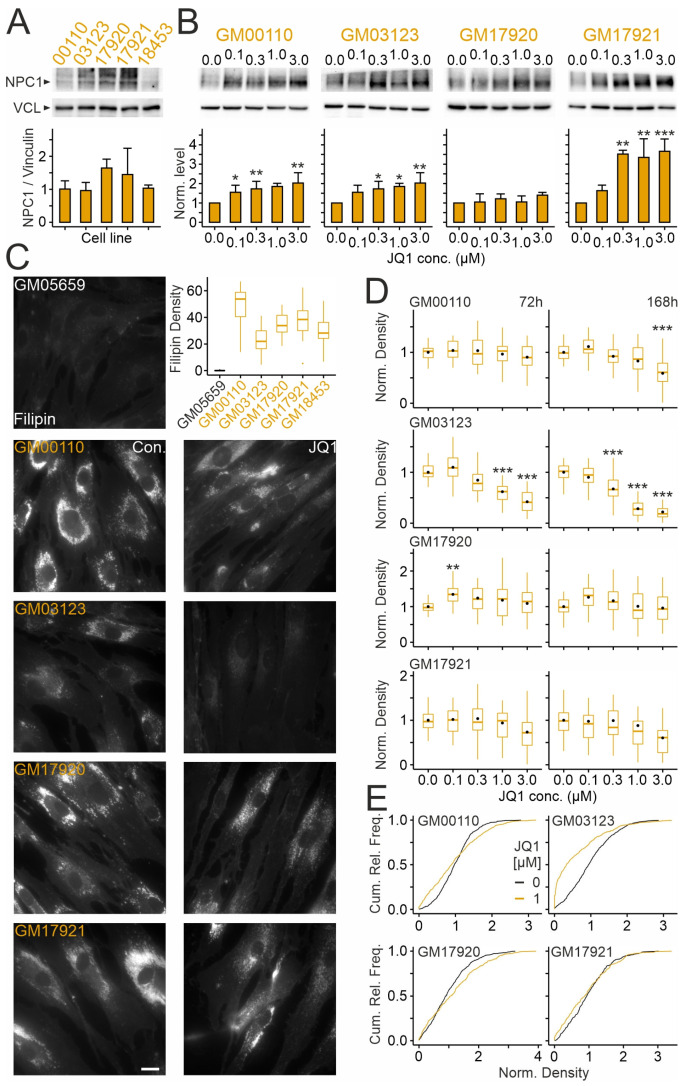 Figure 6
Figure 6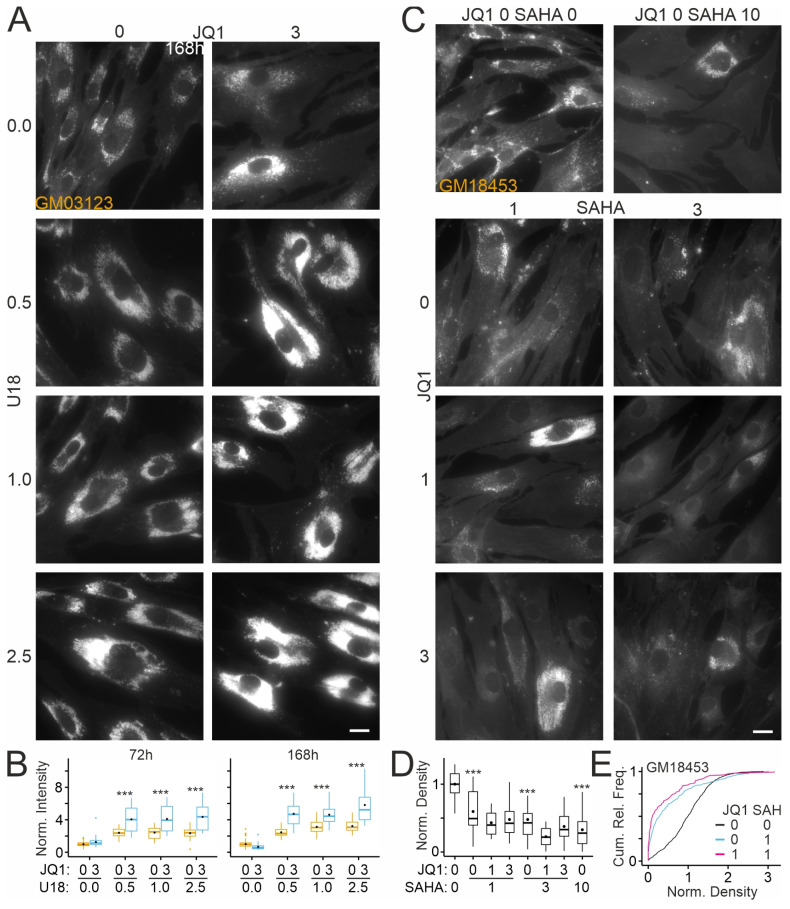 Figure 7
Figure 7- —Centre National de la Recherche Scientifique
- —Université de Strasbourg
- —Niemann–Pick Selbsthilfegruppe e.V.
- —Fondazione Telethon Italy
- —Together Strong Niemann–Pick foundation
- —Ara Parseghian Medical Research Fund
- —BILD Hilft e.V.
- —Rome Technopole
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLysosomal Storage Disorders Research · HIV Research and Treatment · Autoimmune and Inflammatory Disorders Research
1. Introduction
Lysosomal handling of lipids is essential for cellular function [1], and genetic defects in these processes provoke fatal disorders presenting highly variable onset, diverse neurovisceral symptoms, and reduced life spans [2]. A prime example is NPCD, a rare, autosomal-recessive, and pan-ethnic lysosomal disorder presenting progressive and ultimately fatal neurovisceral symptoms [3,4]. Several forms of NPCD are discerned based on the onset of neurologic disease [5,6,7]. Disease incidence is estimated at 1:100,000 [3].
The primary cause of NPCD is specific alleles of the gene NPC intracellular cholesterol transporter 1 (NPC1; OMIM #257220; 95% of cases) [8,9] or the gene NPC intracellular cholesterol transporter 2 (NPC2; OMIM 607625; 5% of cases) [10]. The encoded proteins NPC1 and NPC2, which are ubiquitously expressed, reside in the membrane [11,12] and the lumen of late endosomes [10,13,14], respectively. Phenotypes of mouse models [15], biochemical assays [16,17], and structure analyses [18,19,20,21,22,23] indicate that the two proteins export unesterified cholesterol from the endosomal–lysosomal system, although other functions are discussed. Dysfunction of either protein causes intracellular accumulation of unesterified cholesterol [24,25] and of other lipids [26,27,28,29,30], and impairs lysosomal [29,31,32,33] and mitochondrial function [34], and autophagy [35].
Despite considerable efforts [36], therapeutic options for NPCD are limited to symptomatic treatment and to the disease-modifying drugs N-butyl-deoxynojirimycin (OGT918, Miglustat, Zavesca) [37], arimoclomol (Miplyffa) in combination with Miglustat [38], and N-acetyl-L-leucine (Levacetylleucine) [39].
Here, we explored BET proteins as new drug targets for NPCD. BET proteins control gene expression in a complex manner. They recognize and bind to specific acetyl-lysine patterns on histones, control chromatin structure, and interact with transcription factors to promote or repress transcriptional programs [40,41]. They are considered therapeutic drug targets [42] for different types of cancer [43] and other pathologic conditions [44,45] including cachexia [46], Duchenne muscular dystrophy [47], Fabry disease [48], and retinal inflammation [49]. Recently, we discovered that JQ1, a well-characterized competitive inhibitor of BET proteins [50], enhances the protein content of NPC1 in cultured cells [51]. This effect is in line with evidence that BET proteins control lysosome- and autophagy-related genes in various cell types and disease conditions [47,52,53,54,55,56,57,58,59]. Thus, inhibition of BET proteins may have therapeutic potential in NPCD because disease severity seems to correlate with cellular levels of NPC1 protein [60,61,62] and because some pathogenic variants of NPC1 are functional, but degraded due to misfolding [63,64,65,66,67,68].
Using patient-derived skin fibroblasts as a preclinical model for in vitro drug tests [69], we observed that inhibition of BET proteins by JQ1 increased protein levels of NPC1 and affected pathologic changes in a dose-, time-, and patient-dependent manner. The drug induced an enhancement of lysosomal release, followed by an increase in cholesterol accumulation, and a subsequent decrease after long-term treatment. Effects of JQ1 on cholesterol accumulation were abolished by an NPC1 inhibitor, and varied across fibroblasts from distinct patients. Together, our results support further exploration of BET proteins as new therapeutic targets for NPCD and as new components regulating lysosomal function and cholesterol metabolism.
2. Results
We explored BET proteins as new therapeutic drug targets for NPCD using the membrane-permeant inhibitor JQ1 and primary cultures of dermal fibroblasts from NPCD patients and a healthy donor.
2.1. Effects of JQ1 on Viability, Protein Levels of NPC1, and Its Distribution in Human Skin Fibroblasts
First, we tested whether the drug affects the number and viability of fibroblasts. As shown in Figure 1A, patient-derived fibroblasts carrying the I1061T variant attained lower numbers and showed a lower fraction of propidium iodide-positive (dead) cells compared to fibroblasts from the healthy donor. JQ1 inhibited cell growth and enhanced the percentage of dead cells compared to vehicle (dimethylsulfoxide; DMSO) in a dose- and time-dependent manner, regardless of the genotype (Figure 1B). Anti-proliferative and toxic effects of JQ1 at high doses were reported in dermal fibroblasts [70] and in immortalized cell lines [50,71].
Next, we tested how JQ1 affects NPC1 protein levels in human skin fibroblasts. These experiments followed up on our previous observation that the drug modifies components mediating lipid homeostasis, including NPC1, in a hepatocarcinoma cell line [51]. Immunoblotting revealed lower levels of the I1061T variant compared to the normal version of NPC1 in fibroblasts (Figure 2A), in line with previous studies [63,72,73]. JQ1 enhanced protein levels of NPC1 in a concentration-dependent manner compared to vehicle-treated (JQ1 concentration zero) cultures independently from the genotype, with robust changes occurring after 72 h of treatment (Figure 2B). The fraction of endoglycosidase H (EndoH)-resistant NPC1 protein was unaffected by JQ1 regardless of the variant (Figure 2C), indicating that JQ1 does not modify protein glycosylation under our experimental conditions.
Next, we asked whether JQ1-induced NPC1 protein reaches the endosomal–lysosomal system using double immunocytochemical staining with antibodies against NPC1 and against the endosomal–lysosomal marker LAMP2 (Figure 3A). Quantitative analyses of regions of interest (ROIs) confirmed lower levels of NPC1 and of its colocalization with LAMP2 in patient-derived fibroblasts compared to healthy donor cells (Figure 3B). Treatment of patient-derived cells with JQ1 for 48 or 72 h did not enhance the density of LAMP2 or NPC1-positive clusters (Figure 3A,C) and only moderately affected the colocalization of NPC1 and LAMP2 (Figure 3C).
2.2. Effects of JQ1 on Lysosomes in Patient-Derived Skin Fibroblasts
We tested next whether JQ1 affects the expansion of the acidic (lysosomal) compartment, a cellular hallmark of NPC1 deficiency, using lysotracker, a pH-sensitive fluorescent dye [74,75,76] (Figure 4A–D). Flow cytometry of lysotracker-labeled fibroblasts revealed a higher cell complexity and increased fluorescence signal in cells carrying the disease-causing variant of NPC1 compared to cells from a healthy donor (Figure 4A–C). Treatment of patient-derived cells with JQ1 reduced the lysotracker signal in a dose-dependent manner regardless of the treatment duration (Figure 4D). This effect may have been due to exocytotic release of lysosomal content [77]. To address this, we used two complementary assays [77,78,79]. First, immunocytochemical staining of LAMP2 in non-permeabilized cells revealed the presence of LAMP2 on the cell surface following fusion of the lysosome with the plasma membrane. Second, detection of hexosaminidase activity in the culture medium revealed the cellular release of lysosomal enzymes (Figure 4E–I). The assays showed enhanced basal LAMP2 surface expression (Figure 4E,F) and extracellular hexosaminidase activity (Figure 4H) in patient-derived fibroblasts compared to fibroblasts from a healthy donor, in agreement with a previous study [80]. Treatment with JQ1 further increased both the LAMP2 surface expression (Figure 4G) and extracellular hexosaminidase activity in a time- and concentration-dependent manner (Figure 4I), suggesting that the drug induces release of lysosomal material.
2.3. Effects of JQ1 on Cholesterol Accumulation in Patient-Derived Skin Fibroblasts
We tested next whether JQ1 affects the accumulation of unesterified cholesterol, another hallmark of NPC1 deficiency, using cytochemical staining with the cholesterol-binding drug filipin [24,81] (Figure 5). Patient-derived fibroblasts showed a higher density of filipin-positive puncta (Figure 5A,B) compared to cells from the healthy donor (Figure 5B). Interestingly, JQ1 increased the density of filipin-positive puncta after 48 h of treatment (Figure 5C) and showed no effects after 72 h (Figure 5C). The increase may have been due to enhanced cholesterol synthesis. However, cellular levels of 3-Hydroxy-3-Methylglutaryl-CoA Reductase (HMGCR), a key enzyme mediating cholesterol synthesis, were unaffected by JQ1 during these treatment periods, as shown by immunoblotting (Figure S1). Closer scrutiny of filipin-stained cells treated for 72 h with JQ1 revealed that the drug induces a dual effect in a given cell population, with the majority showing decreased and 30% of cells showing increased filipin densities (Figure 5D). These divergent effects explained the lack of significance and suggested a time-dependent switch of the JQ1 effect. We treated cells with JQ1 for 168 h and found that long-term treatment reduced the density of filipin-positive puncta in patient-derived fibroblasts (Figure 5C,D), indicating time- and dose-dependent effects of the drug on cholesterol accumulation.
2.4. Effects of JQ1 on Skin Fibroblasts from Different NPCD Patients
We next tested how JQ1 affects NPC1 levels and cholesterol accumulation in skin fibroblasts from four patients bearing different variants and polymorphisms. As shown in Figure 6, these lines showed different basal levels of NPC1 protein and different degrees of cholesterol accumulation under untreated conditions. Treatment with JQ1 for 72 h raised the protein levels of NPC1 in a dose-dependent manner in all but one fibroblast line (GM17920). With respect to cholesterol accumulation, the effects of JQ1 differed markedly between patient-derived lines. The spectrum ranged from a robust decrease already within 72 h in line GM03123 to an apparent lack of response in line GM17920, which showed no increase in NPC1 protein, and in line GM17921 despite a strong increase in NPC1 protein. In the other lines, JQ1 reduced cholesterol accumulation after 168 h of treatment (Figure 6D). Notably, in all lines tested, JQ1 showed dual effects, increasing and decreasing puncta density in subsets of fibroblasts from the same culture preparation (Figure 6E) as shown in GM18453 (Figure 5D). These results indicated that JQ1 raises NPC1 levels in most patient-derived lines. With respect to cholesterol accumulation, subsets of cells responded to JQ1 with increased and decreased densities in a time- and dose-dependent manner, with a net reduction occurring in 3 out of 4 lines after prolonged treatment.
2.5. Effects of JQ1 on Cholesterol Accumulation in Patient-Derived Fibroblasts in the Presence of an NPC1 Inhibitor
Our findings raised the question of whether the reduction in cholesterol accumulation by JQ1 depends on NPC1 activity. To address this point, we applied JQ1 in the presence or absence of 3-beta-[2-(diethylamine)ethoxy]androst-5-en-17-one (U18666A or U18), an inhibitor of NPC1 [82], using the GM03123 line (Figure 6D). As shown in Figure 7A,B, treatment with U18 enhanced the intensity of filipin fluorescence in a dose-dependent manner, indicating residual NPC1 activity in these cells. Co-treatment with JQ1 further enhanced the staining intensity at each U18 concentration tested (Figure 7). These results suggested that JQ1 reduces cholesterol accumulation in an NPC1-dependent manner. JQ1 may also act through NPC1-independent pathways that are insufficient to overcome U18-induced cholesterol accumulation.
2.6. Effects of JQ1 on HDAC Inhibitor-Mediated Reduction in Cholesterol Accumulation in Patient-Derived Fibroblasts
Previous studies revealed that pharmacologic inhibition of HDACs reduces cholesterol accumulation in mouse neural stem cells [83] and human fibroblasts [72,84,85] carrying NPC1 variants. Therefore, we tested whether inhibition of BET proteins acting as histone acetylation readers modifies these effects. In line with previous reports, the HDAC inhibitor suberoylanilide hydroxamic acid (Vorinostat; SAHA) reduced the density of filipin-positive puncta in a dose-dependent manner (Figure 7C,D). Interestingly, similar to JQ1 (Figure 5D), SAHA also induced dual effects with fibroblasts in the same culture well, showing enhanced and reduced puncta densities (Figure 7E). The addition of JQ1 further reduced the density of puncta, but the effect did not reach statistical significance (two-way ANOVA; Figure 7D,E).
3. Discussion
Here, we report effects of BET protein inhibition in a cell culture model of NPCD that encourage further exploration of this approach. We found that a prototypical BET protein inhibitor enhanced the cellular level of NPC1 protein, diminished lysosomal expansion and cholesterol accumulation, and induced the release of lysosomal components in a time- and dose-dependent manner. The effects of JQ1 on cholesterol levels depended on NPC1 activity, and the enhancement of protein levels occurred in most of the patient lines tested, but the extent of cholesterol reduction varied in a line-dependent manner.
Our finding that BET protein inhibition enhances protein levels of NPC1 is in line with previous reports that the promoter region of NPC1 is associated with acetylated histones [86] and that de-acetylation increases gene expression [84]. BET proteins may read these patterns and repress NPC1 production. Other means to enhance cellular NPC1 protein levels in vitro are the inhibition of HDACs [65,68,72,87,88,89], enhanced chaperone activity [63,73,90,91,92,93,94,95,96,97], and reduced protein degradation [63,64,90,92]. These manipulations enhanced the presence of NPC1 variants in the endosomal–lysosomal system [63,64,68,73,89,90,91,94,95,96], and they reduced the intracellular accumulation of unesterified cholesterol [63,64,65,68,72,73,84,85,88,89,90,91,95,96]. There is evidence for synergistic effects of BET protein and HDAC inhibitors in transcription regulation [98] and tumor therapy [99,100], but in NPCD patient-derived fibroblasts, JQ1 did not significantly enhance the SAHA-induced reduction in cholesterol accumulation.
Our observation that the responses to JQ1 are patient-dependent is in line with patient-specific effects of HDAC inhibitors [68,72,89]. The outcome probably depends on the specific activity of NPC1 variants generated from each patient’s allele combinations [63,68,89]. Evidently, so-far-unknown genetic or epigenetic modifiers [101,102,103] may further impact the outcome of JQ1 treatment. The effects of JQ1 seemed to depend on NPC1 activity since JQ1 failed to reduce cholesterol accumulation after pharmacologic inhibition of NPC1 by U18. However, this finding does not exclude that JQ1 also acts through NPC1-independent pathways.
Our observation that at 72 h treatment, JQ1 increased and decreased cholesterol accumulation in cells may explain why JQ1 did not affect cholesterol accumulation in a recent fibroblast-based high-throughput drug screen for NPCD [104]. The time-dependent effects of JQ1 may be due to sequential activation of distinct processes or subtype-specific responses [105]. An initial increase in cholesterol accumulation may be due to enhanced levels of lysosomal components as reported previously [52] and an insufficient integration of NPC1 in the endosomal–lysosomal system. During this period, JQ1 also reduced the lysosomal volume as indicated by lysotracker staining. This effect may have been caused by the immediate release of lysosomal content to the extracellular space, as indicated by two independent assays. Lysosomal exocytosis reduces the extent of cholesterol accumulation due to NPC1 dysfunction, as shown previously in cell lines [78,80,106,107,108], patient-derived fibroblasts [80,109,110,111,112,113], primary retinal neurons [79,114], and in NPC1-deficient mice [115]. The net decrease in cholesterol accumulation after prolonged JQ1 treatment may be caused by additional processes that are affected by BET protein inhibition [116], including a reduction in cholesterol biosynthesis [51,117] and of intracellular lipid levels [51], and an increase in apolipoprotein A [118], which has been explored as therapeutic agent for NPCD [119]. In general, BET protein inhibition affects many transcriptional programs through interactions with acetylated lysines on histones, including a site showing epigenetic marks in NPCD [120]. Notably, a lack of effect of BET protein inhibition in some patient lines does not exclude a therapeutic effect of this approach. Miglustat and arimoclomol, which are approved for the treatment of NPCD, did not reduce cholesterol accumulation in cellular models in vitro [95,121,122], and cerebellar Purkinje cells in NPC1-deficient cats showed cholesterol accumulation following miglustat treatment [123].
Taken together, our results reveal that BET proteins regulate NPC1 levels and thereby impact lysosomal function and cholesterol homeostasis depending on specific protein variants and the genetic background. Our results encourage further studies to evaluate their potential as a therapeutic drug target for NPC disease and their contribution to cholesterol homeostasis and lysosomal function in differentiated cells.
4. Materials and Methods
4.1. Cell Culture and Drug Treatment
Human dermal fibroblasts used in this study were obtained from the NIGMS Human Genetic Cell Repository [Coriell Institute for Medical Research, Camden, NJ, USA]. Most experiments were performed using fibroblasts from a NPCD patient homozygous for a frequent pathogenic allele [GM18453, https://catalog.coriell.org/0/Sections/Search/Sample_Detail.aspx?Ref=GM18453 (accessed on 10 June 2025) male, p.Ile1061Thr p.Ile1061Thr] [124] and from a sex- and age-matched healthy donor [GM05659 https://catalog.coriell.org/0/Sections/Search/Sample_Detail.aspx?Ref=GM05659 (accessed on 10 June 2025): male, 14 months old]. Selected experiments were performed using fibroblasts from heterozygous patients carrying different allele combinations [GM00110 https://catalog.coriell.org/0/Sections/Search/Sample_Detail.aspx?Ref=GM00110 (accessed on 10 June 2025): male, 9 years, p.Pro237Ser p.Phe740_Ser741del [60]; GM03123 https://catalog.coriell.org/0/Sections/Search/Sample_Detail.aspx?Ref=GM03123 (accessed on 10 June 2025): female, 9 years, p.Pro237Ser p.Ile1061Thr [60,125]; GM17920 https://catalog.coriell.org/0/Sections/Search/Sample_Detail.aspx?Ref=GM17920 (accessed on 10 June 2025): female, p.Pro401Thr p.Ile1061Thr [94]; GM17921 https://catalog.coriell.org/0/Sections/Search/Sample_Detail.aspx?Ref=GM17921 (accessed on 10 June 2025): male, 5 years, p.Pro433Leu p.Ile1061Thr [126]]. Cells were cultured in Dulbecco’s modified Eagle medium (DMEM) containing high glucose supplemented with 5% fetal bovine serum, 1% L-glutamine, 1% sodium pyruvate, 1% non-essential amino acids, and 1% penicillin/streptomycin (all Sigma-Aldrich/Merck, Milano, Italy) at 37 °C and 5% CO_2_. All experiments were performed at 60–70% cell confluency and maximally 20 passages. The drugs (+)-JQ1 (#SML1524, Sigma-Aldrich/Merck), U18 [#S9669; Selleckchem, Houston, TX, USA] and SAHA (#SML0061, Sigma-Aldrich/Merck) were added to primary cultures at indicated concentrations after dilution from respective stock solutions (JQ1: 3 mM; SAHA: 1 mM in DMSO; U18: 5 µg/mL in ethanol). Control cultures run in parallel were treated with vehicle (0.1% DMSO or 0.1% ethanol or both in DMEM) for the indicated times.
4.2. Cell Number and Viability
Fibroblasts were cultured in 24-well plates (#833922, Sarstedt, Nürmbrecht, Germany) at 15,000 cells/well and treated as indicated. Cells were detached with trypsine/EDTA (0.05%), resuspended in medium, stained with propidium iodide (2 µg/mL; Sigma-Aldrich/Merck) to label dead cells, and subjected to flow cytometry (CytoFlex, Beckman Coulter, Brea, CA, USA) counting all cells and propidium iodide-positive cells (excitation at 488 nm, emission 585/42 band pass filter).
4.3. Lysate Preparation and Immunoblotting
Fibroblasts were cultured in 6-well plates at 150,000 cells/well and treated as indicated. Cells were lysed in homogenization buffer (sucrose 0.1 M, KCl 0.05 M, KH_2_PO_4_ 0.04 M, EDTA 0.04 M, pH 7.4, with proteinase (1:1000; #P8340; Sigma-Aldrich/Merck) and phosphatase inhibitor cocktails (1:400; #P0044; Sigma-Aldrich/Merck) by sonication [VCX 130 PB, Sonics Materials, Newtown, CT, USA] on ice for 20 sec. Then, samples were spun down at 13,000 rpm for 10 min at 4 °C to remove cell debris. Protein concentrations were assessed by the Bradford method (Sigma-Aldrich/Merci) following the manufacturer’s instructions. For immunoblotting, samples were diluted with Laemmli buffer, boiled for 5 min, and subjected to SDS-PAGE (40 µg of protein/lane). Proteins were transferred to nitrocellulose membranes [Trans-Blot Turbo Transfer System; Bio-Rad Laboratories, Hercules, CA, USA]. Membranes were blocked with fat-free milk (5% in Tris-buffered saline 0.138 M NaCl, 0.027 M KCl, 0.025 M Tris-HCl, and 0.05% Tween-20, pH 6.8) for 1 h at room temperature, exposed to antibodies against NPC1 (1:1000; #NB400-148, Novus Biologicals / Bio-Techne S.A.S. Noyal Châtillon sur Seiche, France) or vinculin as loading control (1:40,000; #V9131, Sigma-Aldrich/Merck) or against HMGCR (1:2000, #ab174830; Abcam, Cambridge, UK) and against tubulin (TUB) as loading control (1:40,000; #T6074 Sigma-Aldrich/Merck) overnight at 4 °C followed by corresponding horseradish peroxidase-conjugated secondary IgG antibodies (Bio-Rad Laboratories) for 1 h at room temperature. Chemiluminescence was visualized using the ChemiDoc MP system (Bio-Rad Laboratories) and analyzed by ImageJ software [Version 8; National Institutes of Health, Bethesda, MD, USA].
4.4. Endoglycosidase H Assay
Fibroblasts were cultured in 6-well plates and treated as indicated. Cells were lysed in homogenization buffer (sucrose 0.1 M, KCl 0.05 M, KH2PO4 0.04 M, EDTA 0.04 M, pH 7.4) by sonication (VCX 130 PB, Sonics Materials) at 4 °C for 20 s, and centrifuged at 12,000 rpm for 10 min at 4 °C to yield total lysate. The endoglycosidase H (EndoH) assay (V4875, Promega Italia; Milano, Italy) was performed following the manufacturer’s instructions. The samples contained EndoH reaction buffer, water, and EndoH enzyme. As a negative control, the EndoH enzyme was replaced by water. All samples were incubated at 37 °C for 6 h, and the reaction was terminated by adding Laemmli sample buffer before immunoblotting.
4.5. Immunocytochemical Staining
Fibroblasts were cultured in 96-well microplates (Black/Clear Flat Bottom Imaging Microplate; #353219, BD Falcon, Thermo Fisher Scientific, St. Leon-Rot, Germany) at 3000 cells/well and treated as indicated. Following treatment, cells were washed three times with phosphate-buffered saline (PBS), and chemically fixed with 4% paraformaldehyde (in PBS) for 15 min at room temperature. Cells were permeabilized (saponin 0.05% in PBS; #84510, Sigma-Aldrich/Merck) for 10 min, and incubated for 45 min with blocking solution (3% bovine serum albumin with 1% goat serum in PBS) and then overnight at 4 °C with primary antibodies (1% bovine serum albumin in PBS) against NPC1 (1:2500; NB400-148, Novus Biologicals) and LAMP2 [1:1000; #sc-18822, Santa-Cruz, Dallas, TX, USA/Clinisciences, Nanterre, France]. After incubation, cells were washed and reacted for 1 h at room temperature with appropriate secondary antibodies (1:1000; goat anti-rabbit secondary antibody Alexa Fluor 546; #A-10040, Thermo Fisher Scientific, St. Leon-Rot, Germany; goat anti-mouse secondary antibody Alexa Fluor 488; #A-11001, ThermoFisher Scientific). Fluorescence was visualized and digitized using an upright microscope (ZEISS Observer 7; Carl Zeiss France S.A.S., Rueil-Malmaison, France/Lordil, Lay-Saint-Chrisophe, France) equipped with a light source (ZEISS Colibri; Carl Zeiss France S.A.S./Lordil), objectives (40× water, N.A. 1.2; 63× oil, N.A. 1.4; Carl Zeiss France S.A.S./Lordil), a module for optical sectioning by structured illumination (ApoTome.2; Carl Zeiss France S.A.S./Lordil) and a digital camera (Hamamatsu ORCA-Flash 4.0; Carl Zeiss France S.A.S./Lordil). Densities of LAMP2- and NPC1-positive puncta were determined in manually outlined regions of interest (ROI) (1–5 per soma) using custom-written LabVIEW (Version 6.0; National Instruments, Austin, TX, USA) routines [79]. Colocalization was estimated based on Pearson’s correlation coefficient of NPC1 and LAMP2 fluorescence intensities in individual puncta detected in ROIs.
4.6. Cytochemical Staining
Fibroblasts were cultured in 96-well microplates (black imaging plate; #353219, BD Falcon, Schaffhausen, Switzerland) at 3000 cells/well and treated as indicated. Following treatment, cells were washed three times with PBS, fixed by paraformaldehyde (4% in PBS) for 15 min at room temperature and stained with filipin (50 μg/mL in PBS prepared freshly from a 250-fold ethanolic stock; #F9765, Sigma-Aldrich/Merck) for 2 h at room temperature in the dark. Fluorescence images of stained cells were acquired using an inverted microscope (Axiovert 135TV; Carl Zeiss Microscopy GmbH, Oberkochen, Germany) equipped with a metal halide lamp (10%; Lumen 200, Prior Scientific Instruments GmbH, Jena, Germany), an appropriate excitation/emission filter (XF02-2; Omega Optical, LLC/Laser Components S.A.S., Meudon, France), a 40× objective (oil, N.A. 1.3; Carl Zeiss Microscopy GmbH) and an air-cooled monochrome charge-coupled device camera (Sensicam, PCO Computer Optics, Kehlheim, Germany) controlled by custom-written LabVIEW routines (Version 6.0; National Instruments). Densities of filipin-positive puncta and fluorescence intensities were determined in 10 to 12 images per condition and preparation from manually outlined ROIs (1–5 per cell) using custom-written LabVIEW routines (Version 6.0; National Instruments) [79].
4.7. Lysotracker Staining and Cytometry
Fibroblasts were cultured in 6-well plates (#833920, Sarstedt) at 150,000 cells/well and treated as indicated. Before the end of the treatment, cells were incubated with LysoTracker Red DND-99 (1 µM in culture medium; #L7528, Life Technologies/Thermo Fisher Scientific) for 30 min at 37 °C, detached, spun down at 13,000 rpm, and resuspended in 300 μL of medium prior to flow cytometry. Data were acquired using a flow cytometer [Cytoflex-LX; Beckman-Coulter, Brea, CA, USA] and analyzed by specialized software [Kaluza Version 2.1, Beckman-Coulter; FloJo Version 10, Becton-Dickinson, Ashland, OR, USA]. Fold-changes in LysoTracker intensity were calculated as ratios of geometric means of stained/unstained samples as described [74].
4.8. Hexosaminidase Activity Assay
Fibroblasts were cultured in 96-well microplates (#833924, Sarstedt) at 4000 cells/well in DMEM without phenol red. The assay was performed similarly as described [79]. Briefly, following treatment, 20 µL of cell culture medium was incubated at 37 °C for 3 h with 20 µL reaction mix containing sodium citrate (10 mM; pH 4.2) and 4-methylumbelliferyl-2-acetamido-2-deoxy-b-D-glucopyranoside (2 mM; #474502, Sigma-Aldrich/Merck). The reaction was stopped by 5 volumes of glycine and Na_2_CO_3_. (0.2 M). Crystal violet (#C0775, Sigma-Aldrich/Merck; 0.05% in H_2_O with 1% paraformaldehyde and 1% methanol) was added to the cell suspension to indicate cell number. The fluorescent product 4-methylumbelliferone and crystal violet were measured in triplicate on a microplate reader (Tecan Spark, Männedorf, Switzerland) using suitable filters (excitation: 365 nm; emission filters: 440 nm; absorbance: 585 nm). Calibration curves were acquired using defined amounts of the fluorescent product 4-methylumbelliferone sodium salt (#M1508, Sigma-Aldrich/Merck). Fluorescence of 4-methylumbelliferone was normalized to crystal violet intensity.
4.9. Data Analysis and Visualization
Data analysis and visualization were accomplished with ImageJ software [version 8, National Institutes of Health, Bethesda, MD, USA], and with custom-written routines using the open-source software R (Version 4.0.5; [127] and selected packages (data.table: version 1.14.0, ggplot2: Version 3.3.3). Unless indicated otherwise, results are displayed using bar and whisker plots representing mean and standard deviation, respectively. Statistical tests were performed as indicated. When comparing three or more experimental groups, analysis of variance (one- or two-way ANOVA) was carried out, followed by Tukey’s post hoc test as indicated. Asterisks indicate statistically significant differences based on p values (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
5. Patents
An Italian patent (No. 102021000015467) has resulted from the work reported in this article.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ebner M. Fröhlich F. Haucke V. Mechanisms and functions of lysosomal lipid homeostasis Cell Chem. Biol.20253239240710.1016/j.chembiol.2025.02.00340054455 · doi ↗ · pubmed ↗
- 2Ludlaim A.M. Waddington S.N. Mc Kay T.R. Unifying biology of neurodegeneration in lysosomal storage diseases J. Inherit. Metab. Dis.202548 e 1283310.1002/jimd.1283339822020 PMC 11739831 · doi ↗ · pubmed ↗
- 3Vanier M.T. Niemann-pick disease type c Orphanet J. Rare Dis.201051610.1186/1750-1172-5-1620525256 PMC 2902432 · doi ↗ · pubmed ↗
- 4Berry-Kravis E. Niemann-pick disease, type c: Diagnosis, management and disease-targeted therapies in development Semin. Pediatr. Neurol.20213710087910.1016/j.spen.2021.10087933892845 · doi ↗ · pubmed ↗
- 5Bolton S.C. Soran V. Marfa M.P. Imrie J. Gissen P. Jahnova H. Sharma R. Jones S. Santra S. Crushell E. Clinical disease characteristics of patients with niemann-pick disease type c: Findings from the international niemann-pick disease registry (inpdr)Orphanet J. Rare Dis.2022175110.1186/s 13023-022-02200-435164809 PMC 8842861 · doi ↗ · pubmed ↗
- 6Yilmaz S.B. Baruteau J. Rahim A.A. Gissen P. Clinical and molecular features of early infantile niemann pick type c disease Int. J. Mol. Sci.202021505910.3390/ijms 2114505932709131 PMC 7404201 · doi ↗ · pubmed ↗
- 7Las Heras M. Szenfeld B. Ballout R.A. Buratti E. Zanlungo S. Dardis A. Klein A.D. Understanding the phenotypic variability in niemann-pick disease type c (npc): A need for precision medicine Npj Genom. Med.202382110.1038/s 41525-023-00365-w 37567876 PMC 10421955 · doi ↗ · pubmed ↗
- 8Carstea E.D. Morris J.A. Coleman K.G. Loftus S.K. Zhang D. Cummings C. Gu J. Rosenfeld M.A. Pavan W.J. Krizman D.B. Niemann-pick c 1 disease gene: Homology to mediators of cholesterol homeostasis Science 199727722823110.1126/science.277.5323.2289211849 · doi ↗ · pubmed ↗