Probing Hydrogen-Bonding Preferences and Methyl Internal Rotation in Sotolon and Sotolon-(H2O)1,2
Andrés Verde, Juan Carlos López, Susana Blanco

TL;DR
This study explores how water interacts with sotolon and its hydrates using microwave spectroscopy and theory.
Contribution
The novel contribution is the detailed investigation of hydration effects on sotolon's conformation and methyl rotation.
Findings
Sotolon's conformation is stabilized by an intramolecular hydrogen bond.
Water forms hydrogen-bonded cycles in hydrated sotolon complexes.
Hydration modulates the methyl internal rotation barrier in sotolon.
Abstract
Sotolon is a chiral furanone derivative featuring three distinct oxygen atoms at carbonyl, hydroxyl, and cyclic ether groups that can serve as hydrogen-bond acceptor sites, making it an ideal model system for probing water’s preferential interactions with competing functional groups. In this study, the rotational spectrum of sotolon and its microsolvated complexes, representing the early stages of hydration, was investigated using chirped-pulse Fourier transform microwave (CP-FTMW) spectroscopy. The conformational landscape of sotolon is dominated by a single conformer stabilized by an intramolecular O–H···O=C hydrogen bond. During hydration, water molecules disrupt this interaction by forming closed hydrogen-bonded cycles, resulting in mono- and dihydrated complexes. High-level theoretical calculations underscore the central role of electrostatic interactions in stabilizing these…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
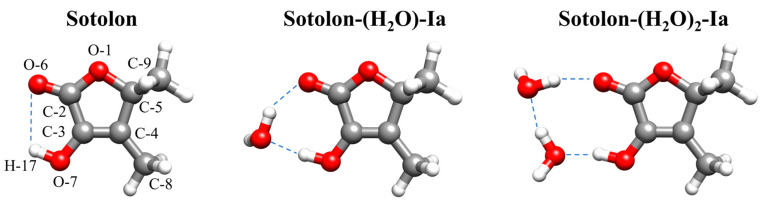 Figure 1
Figure 1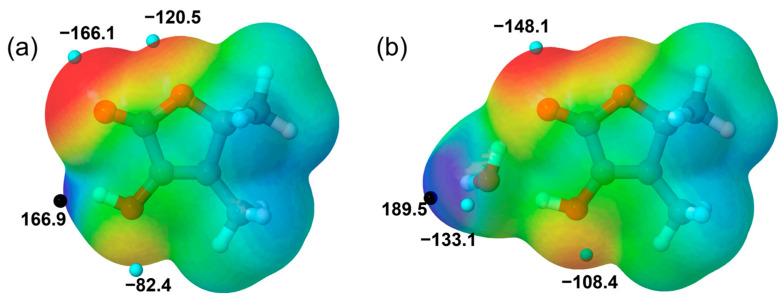 Figure 2
Figure 2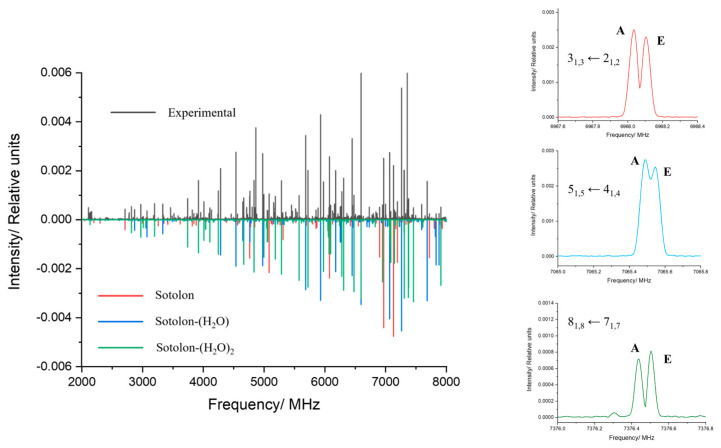 Figure 3
Figure 3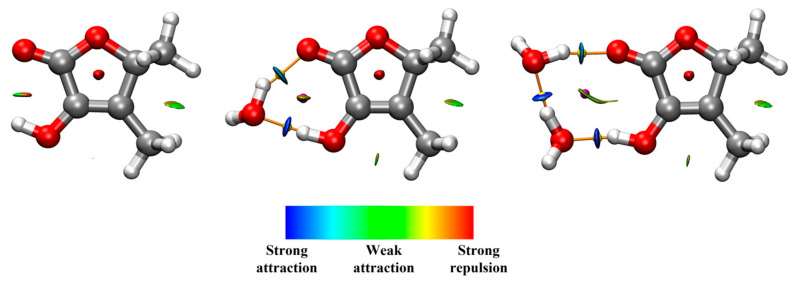 Figure 4
Figure 4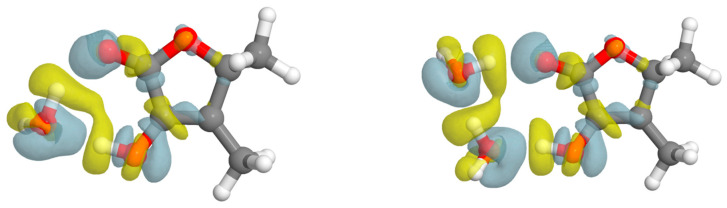 Figure 5
Figure 5- —Junta de Castilla y León
- —Ministerio de Ciencia e Innovación
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMolecular Spectroscopy and Structure · Advanced Chemical Physics Studies · Crystallography and molecular interactions
1. Introduction
Hydrogen bonding (HB) is one of the most extensively studied types of non-covalent interactions, owing to its ubiquity in both chemistry and biology. Its significance is particularly evident in processes such as protein folding, structural stabilization of proteins, and molecular recognition, among many others [1,2,3]. These weak interactions occur predominantly in the solid and liquid phases and play a crucial role in the crystal packing of organic compounds, as well as in the three-dimensional architecture of biological macromolecules [4,5]. Due to their dual role as proton donors and acceptors, water molecules form extensive hydrogen-bonding networks that predominantly govern their interactions with solutes.
The investigation of small clusters involving water molecules, commonly referred to as microsolvation, is of considerable biological and chemical interest, as it enables a deeper understanding of the strength and nature of HBs and other intermolecular interactions. Moreover, the study of microsolvates is particularly valuable for examining the stepwise hydration of organic molecules. This approach allows researchers to determine the conditions under which droplet-like aggregation, where water molecules form self-associated networks resembling those in pure water clusters, is favored over the wetting pathway, where significant deviations from the behavior of pure water clusters are observed [6]. These molecular clusters can be generated and isolated in a controlled manner within the cold environment of supersonic jet expansions and subsequently detected using high-resolution spectroscopic techniques.
Rotational spectroscopy has been widely employed to investigate clusters containing increasing numbers of water molecules, thereby revealing the remarkable adaptability of water to various solutes [7,8,9,10]. Owing to the high structural resolution of this technique, several notable features have been observed in different hydrates, particularly those arising from cooperative effects, as exemplified by formamide–(H_2_O)n complexes [11]. Given the significance of chirality in biological systems, microsolvated clusters of chiral molecules have also been characterized using this method [12]. In this context, recent advances in rotational spectroscopy, such as three-wave mixing and chiral tagging, have enabled the detailed investigation of chiral molecules in the gas phase with unprecedent sensitivity and specificity [13,14]. For example, the combined application of the enantioselective synthesis and analysis of isotopically chiral molecules through chiral tag rotational spectroscopy enables precise determination of absolute configuration and enantiomeric excess without reference samples, offering a robust framework for studying non-covalent interactions in gas-phase systems [14].
In addition, rotational spectroscopy enables the analysis of intramolecular dynamics in the detected systems, including methyl internal rotation. This motion induces characteristic splittings in the rotational spectrum, known as A/E splittings, arising from the interaction between overall molecular rotation and the torsional motion of the methyl group. The magnitude and pattern of these splittings provide direct insight into the internal rotation potential and allow for the accurate determination of the methyl torsional barrier [12,15]. Studies of methyl carbamate complexes with up to three water molecules reveal an increase in the internal rotation barrier with increasing hydration [15], a trend also observed for methyl lactate and its microsolvates in the gas phase [16]. Conversely, in the case of 3-methylcatechol, the addition of up to five water molecules has only a minimal effect on the methyl internal rotation barrier [6,17]. Interestingly, in some systems, such as 4-methylthiazole, complexation with a single water molecule has been found to slightly decrease the barrier height compared to the monomer [18].
To investigate the preferential binding of water to distinct functional groups and to gain deeper insight into the influence of microsolvation on methyl internal rotation, sotolon was selected as an exemplary model system. Sotolon (3-hydroxy-4,5-dimethylfuran-2(5H)-one) is a lactone derivative of 2-furanone, characterized by three distinct oxygen atoms corresponding to cyclic ether, carbonyl, and hydroxyl functionalities (see Figure 1). This chiral furanone is recognized as a key flavor constituent in coffee, raw cane sugar, and dry white wine, among other matrices [19,20]. Its aromatic profile is exploited industrially due to its notable organoleptic properties and exceptionally low odor thresholds [21,22]. The S-enantiomer of sotolon, which exhibits a substantially lower perception threshold, predominantly contributes to the characteristic aroma of prematurely aged white wines [23]. Given its chirality, sotolon has been the subject of several experimental investigations employing vibrational circular dichroism and optical rotation techniques [22,24]. Furthermore, theoretical studies have demonstrated that sotolon possesses high activation barriers for enol/keto tautomerism processes [24]. A detailed elucidation of the molecular structure of sotolon and its interactions with other species is essential for understanding its specific binding mechanisms with olfactory receptors. To date, rotational spectroscopy has not been applied to the study of sotolon. Nonetheless, structural characterizations of analogous compounds, such as 2-acetylfuran and 2-methyltetrahydrofuran-3-one, have been reported [25,26]. Notably, for 2-acetylfuran, two conformers have been identified, both exhibiting methyl internal rotation with experimentally determined V3 barriers of 320 cm^−1^ and 230 cm^−1^, respectively [25]. The effect of microsolvation on the V3 barriers has been previously reported for molecules such as 4-methylthiazole [18], methyl carbamate [15], methyl lactate [16] and 3-methyl catechol [6]. Such literature data provide a basis to contextualize and complement the findings reported here.
In this study, we present a combined investigation using high-resolution rotational spectroscopy and quantum chemical calculations to elucidate the molecular structure of sotolon and its mono- and dihydrate complexes. The nature and strength of the potential hydrogen bonds are assessed through comprehensive molecular interaction analyses. Due to the presence of methyl groups, effects arising from internal rotation were observed in all detected species. The intramolecular dynamics of these methyl groups and the impact of microsolvation on their internal rotation barriers are also discussed. Thus, the novelty of this work lies in the accurate experimental and computational characterization of water binding preferences to an organic molecule presenting different functional groups, as well as in the analysis of how microsolvation influences the V3 barrier of a methyl group. Owing to the presence of a C_3_ chiral center, sotolon exists as both R and S enantiomers; however, within the scope of this investigation, both enantiomers exhibit identical spectroscopic properties. To avoid ambiguity, the S-enantiomer is used consistently throughout the text and figures.
2. Results and Discussion
2.1. Conformational Panorama
The conformational landscape of sotolon is dominated by a conformer stabilized by an intramolecular HB between the hydroxyl group and the carbonyl oxygen atom. A second conformer was predicted to lie 2030 cm^−1^ higher in energy according to the B3LYP-D3/6-311G++(d,p) level calculations (see Figure 1 and Figure S1 and Table S1).
Prior to examining the conformational panorama of the potential microsolvated complexes of sotolon, it is valuable to investigate the properties of the isolated most stable conformer. The molecular electrostatic potential surface (MEP) reveals that, due to the presence of different functional groups, one local maximum and several minima appear (see Figure 2a). The electrostatic potential maximum corresponds to the region of low electron density surrounding the hydroxyl hydrogen atom. In contrast, the more negative regions, indicative of high electron density, are localized around the oxygen atoms of the three distinct functional groups present in sotolon, each one with a different value.
In accordance with the MEP topology, the most stable monohydrates are associated with the maxima of 166.9 kJ mol^−1^. These conformers, herein referred to as sotolon-H_2_O Ia and Ib, are stabilized by the formation of a strong HB from the sotolon hydroxyl group to the oxygen atom of the water molecule (see Figure 1 and Figure S2). Furthermore, water closes a cycle with the solute through a second HB from water to the carbonyl group. Thus, water disrupts the intramolecular HB stabilizing sotolon. Both forms are practically isoenergetic and differ in the side of the ring plane to which the non-bonded water hydrogen atom is pointing (see Table S2 and Figure S2). Despite almost equal energies, these two conformers are non-equivalent due to the lack of symmetry in sotolon. The different orientation of water causes the µc component of the electric dipole moment to have opposite signs in both conformers. The predicted interconversion barrier between both complexes is 270 cm^−1^ (see Figure S4). The most stable monohydrate, lying 2131 cm^−1^ above, involves the water molecule interacting with the cyclic ether group and the nearest methyl group. In the remaining higher-energy conformers, the water molecule interacts either with two oxygen atoms simultaneously or with other functional groups, but these interactions are less favorable for stabilizing the monohydrated complex (see Table S2 and Figure S2). Monohydrates of the sotolon-2 conformer were not explored, as its high energy makes it unlikely to be populated in the molecular jet and therefore not detectable under the experimental conditions.
The MEP topology of sotolon-H_2_O also shows different maxima and minima as occurs for the monomer (see Figure 2b). Accordingly, the conformational landscape of the dihydrates is similarly dominated by a group of close-in-energy conformers, in which a water dimer forms a cycle with the hydroxyl and ketone groups through the formation of three HBs (see Figure 1). Variations in the arrangement of the two non-bonding hydrogen atoms of water result in four conformers. These forms are herein referred to as sotolon-(H_2_O)2 Ia, Ib, Ic, and Id. The next most stable dihydrate is predicted to lie 976 cm^−1^ higher in energy (see Table S3 and Figure S3).
For all conformers of the three systems studied, the MP2/6-311++G(d,p) level of theory confirms the significant stabilization of the lowest-energy conformers in each case predicted by DFT calculations. Moreover, the rotational constants predicted by both methods are in close agreement (see Tables S4–S6 for MP2 results).
2.2. Rotational Spectrum
The 2–8 GHz CP-FTMW spectrum of the Ne-sotolon-water mixture is shown in Figure 3. It shows sets of strong a-type and moderate b-type rotational transitions that were easily identified from the predicted rotational transitions of the most stable sotolon conformer. Each rotational transition splits into a doublet attributable to the A and E states of the internal rotation motion of a methyl group (see excerpts in Figure 3) with a reasonable low internal rotation barrier. The S/N observed do not allow the observation of the spectra of any heavy-atom isotopologue in natural abundance.
The most stable monohydrated conformers are predicted to be close to a prolate rotor with a value of the Ray asymmetry parameter [27] of −0.67 with a dominant µa dipole moment component but with reasonably high values of µb and µc. The identification of the a-type spectrum of this species in the experimental spectrum was not difficult using the predicted rotational constants. Based on the initial set of observed rotational constants, the assignment of the b-type spectrum was straightforward. However, no evidence of c-type transitions was found. As in the case of the monomer, each transition appears as a doublet, attributed to A/E splittings (see Figure 3). The two low energy predicted conformers, which differ in the orientation of the non-bonded hydrogen atom of the water molecule, exhibit similar rotational constants that would result in a closely spaced spectrum. The potential energy function (Figure S4) predicts an interconversion barrier of 270 cm^−1^, Given the non-equivalence of the hydrate conformers, one might expect conformational relaxation from the higher-energy form to the global minimum during the supersonic expansion. But this is not compatible with the fact that no c-type spectrum has been observed. The calculated barrier height and the fact that the interconversion pathway essentially involves the rotation of H_2_O around the hydrogen bonded O-H bond that are hydrogen-bonded to sotolon, with a small vibrational mass, suggest that the ground vibrational state lies above the interconversion barrier. As a result, the vibrational wavefunction samples a wide range of rotation angles, and the expectation value relevant to the observed spectrum corresponds to an almost coplanar arrangement of the water molecule and sotolon ring, leading to a µc close to zero. As a result, only the spectrum of a single conformer with a- and b-type rotational transitions is observed. In consequence, the split lines were also fitted using the appropriate Hamiltonian for internal rotation.
When removing the measured transitions belonging to the sotolon hydrate, a survey of the spectrum shows the typical patterns corresponding to the a-type spectrum of a prolate rotor. Observed at full resolution, the new lines also show splittings attributable to the A and E states of a methyl rotor. The preliminary set of rotational constants derived from the observed spectrum supports its assignment to the most stable dihydrate, as evidenced by the close agreement between experimental and theoretical values.
The observation of only a-type transitions is consistent with the predicted large µ_a_ dipole moment component and the comparatively small values of µ_b_ and µ_c_ (see Table S3), as calculated at the DFT level.
For each detected form, two independent fits for the A and E states were performed to the A-semirigid rotor Hamiltonian of Watson [28] in the I^r^ representation using Pickett’s SPFIT program [29]. In the analysis of the E states, the effective Hamiltonian was complemented with an additional term ( ), which is defined as
The results for the three observed rotamers are collected in Table 1. According to a perturbational treatment of the methyl group internal rotation problem [30,31] the differences between the rotational constants of the A and E states and the parameters D_a_, D_b_ and D_c_ can be written as
where F is the reduced rotational constant, related to the methyl group inertial moment Iα by
where λg (g = a,b,c) are the direction cosines giving the orientation of the internal rotation axis in the principal inertial axis system, ρg are the components of the vector ρ, defined in terms of the internal rotor moment of inertia Iα, and the molecular moments of inertia Ig as
WE^(1)^ is the first order perturbation coefficient for the E state, whereas WA^(2)^ and WE^(2)^ are the corresponding second order perturbation coefficients for the A and E states, respectively. All these parameters can be calculated from the molecular structure. Those corresponding to the methyl groups are given for the detected species in Table S8. The perturbation coefficients can be taken from the tables of Herschbach [32] where they are given as a function of the reduced barrier:
In this way, it is possible to relate the values of the rotational constants or the parameters Da, Db, and Dc with the reduced barrier s to estimate the value of the barrier V3.
For the three detected species, Da and Db were determined for the E state, whereas Dc has to be fixed to zero. The non-determination of Dc is in good agreement with values of ρc close to zero. Furthermore, the determined values are only consistent with the values of the ρ for the methyl group linked to the sp^2^ carbon atom in position 5 (see Figure 1 and Table S8). From the Da and Db parameters, the values estimated are s = 22–23 and values of V_3_ = 260–280 cm^−1^. Since the differences between the C rotational constants are very small, only the differences between the rotational constants A and B have been used to estimate values of s = 29–33 giving barriers V_3_ = 350–400 cm^−1^.
From those estimations, we have proceeded to fit both A and E states of each species simultaneously using the XIAM program [33] in order to obtain more accurate V3 barrier values. Initial values of methyl top inertial moment Iα, the reduced rotational constant F, and the geometrical parameters describing the orientation of the methyl top were calculated from the available structures (see Table S8). This type of fit requires a preliminary calibration step in order to determine which parameters could be reliably fitted. The final fits determined using both SPFIT and XIAM packages (http://info.ifpan.edu.pl/~kisiel/prospe.htm#table_of_programs, accessed on 25 May 2025) are shown in Table 1. The barriers obtained from the XIAM fit seem to be in good agreement with the barriers estimated from the perturbation treatment using the differences between A and B rotational constants (Equation (3)). All the observed frequencies that have been used for both SPFIT and XIAM fits are given in Tables S12–S20.
2.3. Molecular Structure and Molecular Interactions
Considering the good agreement between the experimentally determined rotational constants and those calculated using DFT methods (B3LYP-D3/6-311++G(d,p)), we can assume that the predicted structures accurately reflect the experimental geometries. From the experimental data shown in Table 1, it can be observed that the rotational constant A remains similar for both the monomer and the monohydrate, suggesting that the water molecule is located near the a-inertia axis of sotolon. Additionally, the values of the planar moment P_b_, which quantifies the mass distribution out of the ac plane, are also relatively similar for both species, indicating that water lies close to the ac plane. Furthermore, the Pc values for the monomer, monohydrate, and dihydrate are all very close, placing water molecules near the ab plane of the molecule, which is nearly coincident with the plane of the sotolon ring.
It is well-established that a high HB strength is associated with short donor–acceptor distances and specific angular orientations. For instance, in the case of carbonyl functional groups, optimal C=O···H_w_ angles should approach 120°, as a consequence of the position of oxygen lone pairs. Consequently, an estimation of the HBs can be made using the DFT geometries (see Tables S9–S11), as these structures can be taken as a reasonably good prediction due to their close alignment with the experimental rotational parameters. The intramolecular interaction that stabilizes sotolon is ca. 2.413 Å and thus, it can be classified as a weak HB [2]. As previously mentioned, water disrupts this interaction in the monohydrate. When water acts as a HB acceptor, the O_7_H_17_···H_w_ distance is 1.733 Å and the angle formed is 173.96°, indicating nearly optimal linearity of the interaction. On the other hand, when water acts as a proton donor, the C_2_=O_6_···H_w_ is 1.854 Å and the angle is 121.14°. The analysis of the dihydrate reveals σ-bond cooperative effects. The O···O distance between water molecules is 2.721 Å, which is notably shorter than the experimental 2.98(4) Å of the isolated water dimer [34]. Furthermore, the O_7_H_17_···H_w_ and C_2_=O_6_···H_w_ distances are in this case predicted to be 1.711 Å and 1.812 Å, respectively. Nevertheless, the direccionability of the HBs is less favorable, exhibiting values of 164.65° and 159.14°, which can be attributed to the restricted spatial availability between the hydroxyl and the carbonyl functional groups of sotolon that does not allow a better disposition for the water dimer.
In order to gain more insights into the strength of the molecular interactions presented in the systems, NCI and Bader’s QTAIM analysis have been carried out (see Figure 4). Although the NCI plot shows a weak attractive interaction between the hydroxyl group and the ketone’s oxygen for the monomer, QTAIM does not show the presence of a (3, −1) bond critical point (BCP) for this interaction. In fact, the O-H···O distance and the angular geometry of this intramolecular contact confirm its weakness.
In the monohydrate, water easily disrupts the weak intramolecular interaction and thus, the complex is strongly stabilized by the formation of a seven-membered pseudo ring through the establishment of two HBs between water molecules and sotolon. QTAIM analysis revealed the existence of the BCPs corresponding to two HBs and the formation of a cycle with a (3, +1) ring critical point (RCP). In order to obtain a more quantitative view of the strength of the interactions, we have employed the relationship between the interaction energy and the electron density ρ(r) at a BCP proposed by Emamian et al. [35]. The interaction energies calculated for the two HBs of this complex are −35.0 kJmol^−1^ for the HB in which the hydroxyl group is involved and −25.7 kJmol^−1^ for the HB established between water and ketone.
According to the dihydrate structure, the HBs established between water molecules and sotolon form a nine-membered pseudo ring. The interaction energy for the interaction between water and the hydroxyl group is −36.8 kJmol^−1^. The fact that this strength is higher than in the monohydrate is consistent with σ-bond cooperativity. The strength of the HB between water molecules is −32.3 kJmol^−1^, while the interaction energy for the HB between water and the ketone’s group has a value of −24.2 kJmol^−1^.
NBO theoretical calculations provide a quantitative point of view of the mentioned interactions. This analysis reflects the intramolecular interaction in the sotolon monomer predicting an small electron density transfer from the O_6_ lone pair to the O_7_-H_17_ antibonding σ^^ orbital resulting in a second order E^(2)^ perturbation energy of 2.30 kJ mol^−1^. For the O-H-O_water_ interaction in sotolon-(H_2_O), the electron density transfer from the water oxygen lone to the hydroxyl O-H σ^^ antibonding orbital gives a second order E^(2)^ energy of 85.20 kJ mol^−1^. On the other hand, the second order E^(2)^ energy for the O_water_-H···O=C HB interaction delocalizating the electrondensity of the carbonyl lone pair into the O-H σ^*^ orbital of water is 30.92 kJ mol^−1^. When evaluating the dihydrate, NBO shows an electron density transfer with E^(2)^ = 93.47 kJ mol^−1^ for the HB between the hydroxyl group and water, E^(2)^ = 68.58 kJ mol^−1^ for the HB interaction between water molecules and E^(2)^ = 5.94 kJ mol^−1^ for the HB interaction between water and the carbonyl oxygen.
BSSE corrected dissociation energies of the detected systems calculated using the counterpoise method [36] are shown in Table 2. The positive value of the interaction energy represents the equilibrium dissociation energy D_e_. Table 2 also shows the average energy per HB, that has been calculated as D_e_/n_HB_, where n_HB_ is the number of HB present in the complex. Comparison between the D_e_/n_HB_ value for the mono- and dihydrate show an increasing average energy per bond energy with the addition of the second water molecule, an indication of σ-bond cooperativity. The values found are also in agreement with the average of the HBs’ energies estimated from the QTAIM results using the equation proposed by Emamiam et al.
The interaction energy can be decomposed into its constituent physical components, facilitating a more profound comprehension of the factors that play key roles in the intermolecular interactions. We have employed the sobEDAw strategy [37], designed for dispersion corrected DFT calculations, to understand the main forces at play in the interactions between sotolon and water (see Table 3). The results found are also consistent with those obtained using SAPT(0)/jun-cc-pVDZ (see Table S7). ΔE_int_, which is the interaction energy, is close to the complexation energy (−De) value found form the BSSE calculation. As it was expected, the major contribution at play is of electrostatic nature, which comes from the donor–acceptor character of water molecules that interconnect two sotolon moieties through the formation of HBs. This predominance of electrostatic forces is also observed in the dihydrate, in which the total energy is higher due to the presence of the three HBs.
The electron density shifts (EDS) [38] maps allow the visualizations of the changes in the electron density of a given complex upon complexation. For both microhydrates, the plot clearly shows the loss (yellow) and gain (blue) regions in the space of the functional groups participating in the HB interactions (see Figure 5). It is worth noting that even the oxygen ester of sotolon, which is not directly involved in establishing HBs with water, suffers some redistribution of charge. Thus, it is to some extent affected by the interactions of sotolon with water. This phenomenon can also be observed in the non-bonded hydrogen atoms of water.
2.4. Methyl Internal Rotation
The observed splittings in the spectrum have been attributed to the methyl internal rotation of one of the two methyl groups of sotolon. The V3 barrier for the methyl internal rotation of the methyl group linked to the sp^3^ hybridation C_5_, has been calculated to be 1214 cm^−1^ at B3LYP-D3/6-311++G(d,p) level of theory. This value is predicted to remain relatively constant with the addition of water, leading to barriers of 1212 cm^−1^ and 1196 cm^−1^ for the mono- and dihydrate, respectively. These predicted interconversion barriers are high enough to quench the observation of A/E splittings in the ground vibrational state rotational spectrum.
Conversely, the DFT values of the V3 barriers calculated for the C_8_ methyl group linked to sp^2^-hybridized C_4_ are 326 cm^−1^, 294 cm^−1^ and 298 cm^−1^ for the monomer, the monohydrate and the dihydrate, respectively. The experimental barriers determined from analysis of the spectra using XIAM program are 372.38(43) cm^−1^, 348.78(24) cm^−1^ and 345.808(74) cm^−1^ respectively. The identification of these barriers as belonging to this methyl group is also supported by the geometrical parameters associated with this vibration determined experimentally (see Table S8). Note that the theoretical barrier values are lower than the experimental ones; however, they effectively replicate the trends of the methyl internal rotation barrier upon successive addition of water to sotolon in the mono and dihydrate. The addition of the first water molecule has the effect of reducing the value of the interconversion barrier by approximately 30 cm^−1^. However, the addition of a second water molecule does not result in a significant alteration in the value of the barrier. The comparison between the theoretical and experimental interconversion barriers is shown in Table 4. The observed effect of the water addition could be a consequence of the redistribution of charge resulting from the formation of HBs formed between water and sotolon. This behavior is analogous to that observed for 4-methylthiazole [18] but contrary to those of methyl carbamate [15] or methyl lactate [16]. The fact that the addition of the second molecule does not affect the value with respect to the monohydrate is also consistent with the effect observed in 3-methylcatechol [6,17].
3. Materials and Methods
3.1. Experimental Details
The rotational spectrum of sotolon was recorded in the frequency range of 2 to 8 GHz with a CP-FTMW spectrometer described elsewhere [39,40]. A commercial sample of sotolon was placed in a heatable reservoir at the nozzle and maintained at 150 °C. The resulting sotolon vapor was seeded in a neon stream at a stagnation pressure of 2 bar. A supersonic jet was generated by the expansion of the gas mixture through a small diameter nozzle into a high-vacuum chamber. Microwave excitation was achieved using multifrequency chirped pulses of 4 μs duration, generated by an arbitrary waveform generator (AWG) and amplified to 200 W with a traveling wave tube (TWT) amplifier. The induced molecular polarization was broadcast by a horn antenna oriented perpendicular to the expansion axis. The subsequent free induction decay (FID) signal was detected by a second horn antenna, digitized using a high-speed oscilloscope, and Fourier transformed to yield the frequency-domain spectrum.
Each expansion was probed with eight acquisition cycles, and the resulting spectra were averaged. The experiment was conducted at a repetition rate of 5 Hz, providing sufficient time for the vacuum chamber to recover between cycles.
For the sotolon–water complexes, water vapor was introduced by placing a small reservoir of liquid water in the neon gas line upstream of the nozzle, enabling co-expansion and efficient microsolvation.
3.2. Theoretical Methods
The conformational preferences of sotolon, sotolon-(H_2_O) and sotolon-(H_2_O)2 systems were investigated with the aid of Conformer-Rotamer Ensemble Sampling (CREST) tool (https://crest-lab.github.io/crest-docs/, accessed on 25 May 2025) [41,42]. All the predicted conformers and complexes were further optimized employing the B3LYP hybrid density functional [43]. with the D3 Grime’s dispersion correction term [44] and the Pople’s 6-311++G(d,p) basis set [45]. This level of theory has been shown to provide reliable predictions of rotational constants for both isolated molecules and molecular clusters, making it particularly suitable for the present investigation [46,47]. Harmonic frequency calculations were also carried out to ensure that all the conformers are indeed minima in the potential energy surface. Furthermore, ab initio MP2 [48] optimizations were also performed for all the potential conformers and complexes. The internal rotation barrier (V3) of both methyl groups was calculated for the detected species using the DFT method. All the calculations were performed using GAUSSIAN16 software package [49]. The maxima and minima in the molecular electrostatic potentials have been calculated on the 0.001 a.u. electron density isosurface with the Multiwfn program (http://sobereva.com/multiwfn/, accessed on 25 May 2025) [50].
Non-Covalent Interactions (NCI) [51], Bader’s Quantum theory of Atoms in Molecules (QTAIM) [52], and Natural Bond Orbitals (NBO) [53] analysis were conducted to provide insights about the nature and strength of the molecular interactions. B3LYP-D3/6-311++G(d,p) calculations were also performed to estimate the dissociation energies of the detected complexes using the counterpoise method to correct the basis set superposition error (BSSE) [36]. Energy decomposition analyses were performed using symmetry adapted perturbation theory (SAPT) calculations [54,55] and alternatively following the sobEDAw method [37] were carried out to obtain information about the nature of the intermolecular interactions that stabilize the complexes using PSI4 (https://psicode.org/psi4manual/master/index.html, accessed on 25 May 2025) [56] and Multiwfn [50] programs, respectively.
Moreover, the electron density shift [38] upon complexation was evaluated as the difference in the electron density of the complex and that of the monomers in the geometry of the complex.
4. Conclusions
In summary, the results presented here highlight the novelty of combining high-resolution rotational spectroscopy and quantum chemical methods to investigate how water selectively interacts with different functional groups of a given organic molecule. In particular, the conformational preferences of sotolon and sotolon-(H_2_O)1–2 have been unveiled. The broadband rotational spectrum reveals that the most stable conformer is stabilized by a weak O-H···O=C intramolecular interaction. The addition of water easily disrupts this interaction, with the water molecule(s) linking the hydroxyl and ketone moieties of sotolon through strong HBs, allowing the formation of a seven-membered pseudo ring in the monohydrate and a nine-membered pseudo ring in the dihydrate. Analysis of intermolecular interactions shows that electrostatic contributions are the most significant. They represent ~65% of the total attractive interactions in both microsolvates.
The observation of A/E splittings in the rotational spectrum and their detailed analysis have allowed us to obtain new information about the internal dynamics of sotolon in the monomer and hydrates. Only the methyl group attached to the sp^2^-hybridized carbon atom exhibits an interconversion barrier low enough to display A/E splittings in the ground vibrational state rotational spectrum. The analysis of the experimental data has allowed us to determine the V3 internal rotation barrier for this methyl group in the detected forms. The addition of the first water molecule and the establishment of two HBs result in a slight decrease in the barrier with respect to the monomer. This might be attributed to the internal electronic change redistribution in sotolon upon complexation. However, the addition of a second water molecule has a clearly less pronounced effect on the value of the barrier. The investigation of large amplitude vibrations, such as methyl group internal rotation, in molecules and their complexes provides a powerful probe that reveals how complexation influences molecular properties that would otherwise remain undetected by standard analytical approaches.
Future experimental and theoretical investigations may reveal whether microsolvated clusters containing three or more water molecules continue to grow in a chain-like trend or as interaction between the monomer and the pure water cluster, whether such extended solvation induces any detectable structural changes in the chiral solute, and how the internal rotation barrier evolves upon further hydration. This work can also set a basis for future chiral tagging investigations.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jeffrey G.A. An Introduction to Hydrogen Bonding Oxford University Press Oxford, UK 1997
- 2Desiraju G.R. Steiner T. The Weak Hydrogen Bond in Structural Chemistry and Biology Oxford University Press Oxford, UK 1999
- 3Martín-Fernández C. Montero-Campillo M.M. Alkorta I. Hydrogen Bonds Are Never of an “Anti-Electrostatic” Nature: A Brief Tour of a Misleading Nomenclature J. Phys. Chem. Lett.2024154105411010.1021/acs.jpclett.4c 0077938634115 PMC 11033937 · doi ↗ · pubmed ↗
- 4Lehn J.-M. Toward Self-Organization and Complex Matter Science 20022952400240310.1126/science.107106311923524 · doi ↗ · pubmed ↗
- 5Busch S. Bruce C.D. Redfield C. Lorenz C.D. Mc Lain S.E. Water Mediation Is Essential to Nucleation of β-Turn Formation in Peptide Folding Motifs Angew. Chem. Int. Ed.201352130911309510.1002/anie.20130765724130065 PMC 4227566 · doi ↗ · pubmed ↗
- 6Hazrah A.S. Insausti A. Ma J. Al-Jabiri M.H. Jäger W. Xu Y. Wetting vs. Droplet Aggregation: A Broadband Rotational Spectroscopic Study of 3-Methylcatechol⋅⋅⋅Water Clusters Angew. Chem. Int. Ed.202362 e 20231061010.1002/anie.20231061037697450 · doi ↗ · pubmed ↗
- 7Burevschi E. Chrayteh M. Murugachandran S.I. Loru D. Dréan P. Sanz M.E. Water Arrangements upon Interaction with a Rigid Solute: Multiconfigurational Fenchone-(H 2O)4–7 Hydrates J. Am. Chem. Soc.2024146109251093310.1021/jacs.4c 0189138588470 PMC 11027134 · doi ↗ · pubmed ↗
- 8Macario A. López J.C. Blanco S. Molecular Structure of Salicylic Acid and Its Hydrates: A Rotational Spectroscopy Study Int. J. Mol. Sci.202425407410.3390/ijms 2507407438612884 PMC 11012204 · doi ↗ · pubmed ↗