Genomic Insights into Basal Diptera Phylogeny: The Non-Monophyletic Nature of Blephariceromorpha
Yaoming Yang, Jiayao Ren, Xuhongyi Zheng, Lingna Cai, Jiayin Guan, Tianlong Cai, Xiaodong Xu, Ying Zhen

TL;DR
This study uses genomic data to clarify the evolutionary relationships of early Diptera insects, showing that Blephariceromorpha is not a single evolutionary group.
Contribution
The study resolves the non-monophyletic nature of Blephariceromorpha using mitochondrial and nuclear genomic data.
Findings
Blephariceridae, Deuterophlebiidae, and Nymphomyiidae are each monophyletic.
Deuterophlebiidae and Nymphomyiidae form a sister group representing the basal-most Diptera lineage.
Blephariceridae is placed within Psychodomorpha, not Blephariceromorpha.
Abstract
Diptera is one of the most ecologically significant and species-rich insect orders, but there are still unresolved phylogenetic relationships among its basal lineages, particularly within the infraorder Blephariceromorpha, due to limited molecular data. To address this gap, this study employs two parallel genomic approaches: mitochondrial genomes and nuclear genomic analysis, covering 64 families and over 100 species of Diptera and their outgroups, to elucidate these phylogenetic relationships. Our results strongly support the monophyly of each constituent family (Blephariceridae, Deuterophlebiidae, and Nymphomyiidae), yet they reject the monophyly of Blephariceromorpha. Crucially, we found that Deuterophlebiidae and Nymphomyiidae form a sister group representing the basal-most lineage of Diptera, whereas Blephariceridae is positioned within Psychodomorpha. This indicates that the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
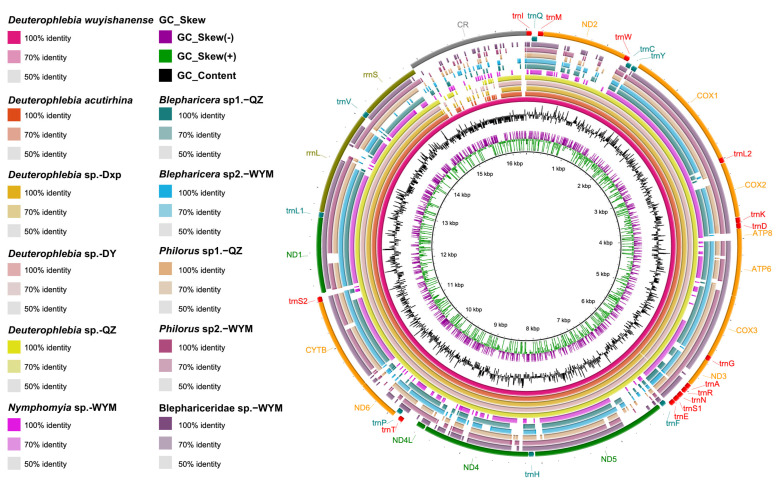 Figure 1
Figure 1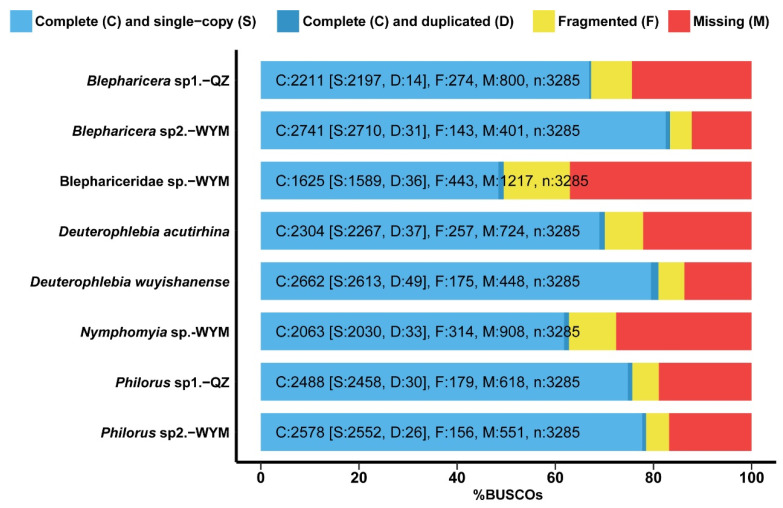 Figure 2
Figure 2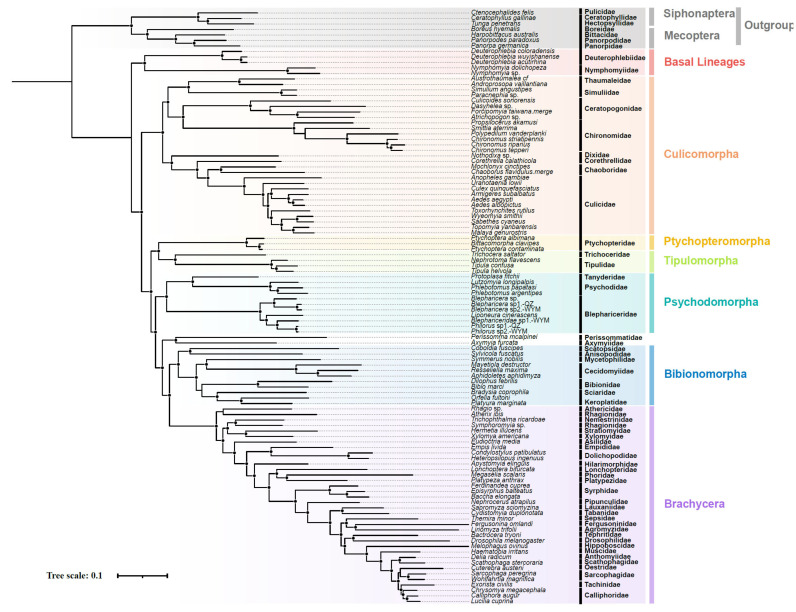 Figure 3
Figure 3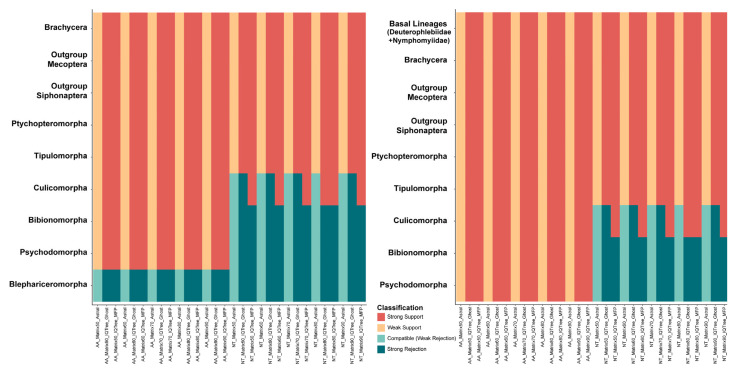 Figure 4
Figure 4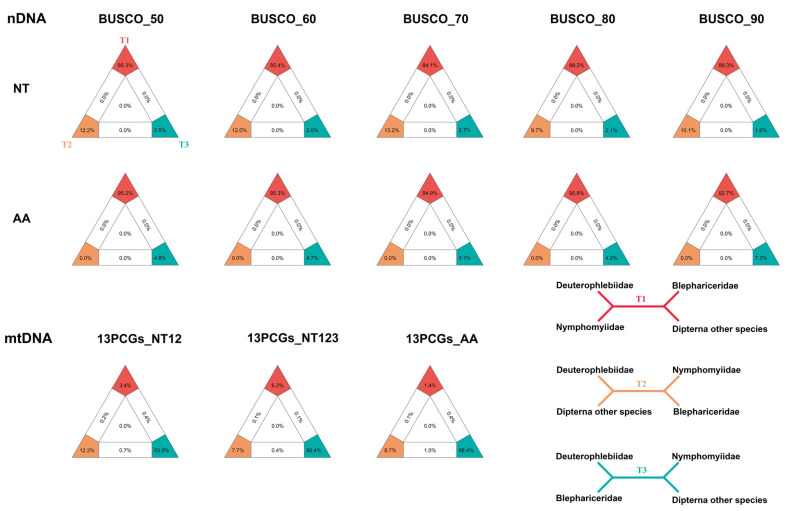 Figure 5
Figure 5- —National Natural Science Foundation of China
- —“Pioneer” and “Leading Goose” R&D Program of Zhejiang
- —Research Center for Industries of the Future (RCIF) at Westlake University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDiptera species taxonomy and behavior · Insect behavior and control techniques · Fossil Insects in Amber
1. Introduction
Diptera, commonly known as true flies, represent one of the largest insect orders, comprising over 165,000 described species [1]. This order traditionally is classified into two primary suborders, the paraphyletic Nematocera (meaning “thread-horns”) includes all flies except those from the monophyly suborder Brachycera (meaning “short-horns”), which encompasses more familiar species such as the housefly and the common fruit fly [2,3,4,5,6]. Nematocera, encompassing approximately 52,000 species distributed across 36 families, is characterized by elongated bodies and long, multi-segmented antennae [1,7]. Species within this suborder exhibit remarkable morphological and ecological diversity, with species occupying virtually every terrestrial and aquatic habitat. Their feeding strategies range from hematophagy, predation, and parasitism to herbivory and saprophagy [8]. Understanding the phylogeny of Nematocera is crucial for elucidating their evolutionary history, interpreting their ecological functions from decomposition to pollination, and assessing their socioeconomic impact, ranging from mosquito-borne diseases to agricultural pest management [9,10,11,12].
Within the systematic classification of Nematocera, the infraorder Blephariceromorpha (comprising Blephariceridae, Deuterophlebiidae, and Nymphomyiidae) has remained one of the most contentious issues in dipteran systematics [2,3,4,5,7,13,14,15,16,17,18,19]. Traditional morphological analyses grouped these families based on shared larval adaptations to fast-flowing streams (e.g., prolegs and suction organs) [13,15,16]. However, conflicting hypotheses have emerged: (1) Nymphomyiidae as sister to Culicomorpha [20], (2) Deuterophlebiidae and/or Nymphomyiidae as the basal dipteran lineage [2,5,14,17], and (3) Blephariceridae as member of Psychodomorpha [2,5,14]. The unstable position of Deuterophlebiidae (alternatively sister to Blephariceridae or Nymphomyiidae) with conflicting phylogenetic evidence exemplifies how specialized habitat adaptations can obscure phylogenetic signals [2,5,7]. Resolving these conflicts requires nuclear genomic-scale data to overcome limitations of fragmentary molecular datasets and morphological homoplasy.
Recent phylogenetic studies utilize diverse molecular datasets, including mitochondrial genomes and nuclear data, to explore evolutionary relationships [21,22,23,24]. Advances in sequencing technology and analytical methods have significantly expanded the availability of nuclear genomic data, yielding extensive collections of phylogenetically informative orthologous loci that enables the resolution of complex evolutionary relationships across wider taxonomic spectra [21,22,24]. The integrated analysis of mitochondrial and nuclear datasets (both transcriptomic and genomic) offers complementary perspectives for resolving deep evolutionary relationships and establishing robust phylogenetic frameworks for insects [25,26,27]. However, data for Blephariceromorpha remain limited, with only one or two species per family represented by mitochondrial or transcriptomic data [14]. This limitation emphasizes the need to supplement existing datasets to robustly investigate the phylogeny of these groups using both mitochondrial and transcriptomic data.
In this study, we present a comprehensive molecular dataset comprising 11 species from the three families within the infraorder Blephariceromorpha (5 Blephariceridae, 5 Deuterophlebiidae, and 1 Nymphomyiidae), with mitochondrial genomes sequenced for all species and transcriptome data obtained for 8 species (Table 1). To address the phylogenetic controversies within Blephariceromorpha, we implemented complementary mitochondrial and nuclear genomic approaches. Our analyses conclusively demonstrate monophyly for each constituent family (Blephariceridae, Deuterophlebiidae, and Nymphomyiidae) but reject the traditional grouping of Blephariceromorpha. Instead, Deuterophlebiidae and Nymphomyiidae form a sister group as the basal lineage of Diptera, while Blephariceridae is placed within Psychodomorpha. These findings enhance our understanding of Nematocera phylogeny and evolutionary diversification within Diptera.
2. Results
2.1. Mitogenomic and Transcriptomic Data Analysis
Our study generated complete or nearly complete mitochondrial genomes of 11 Blephariceromorpha species, including 5 from Blephariceridae, 5 from Deuterophlebiidae, and 1 from Nymphomyiidae, with genome sizes ranging from 16,402 to 16,757 base pairs (bp) (Figure 1 and Table S1). All mitogenomes contained the standard set of 13 protein-coding genes (PCGs), two ribosomal RNA (rRNA) genes, 22 transfer RNA (tRNA) genes, and one control region (CR) (Supplementary Files). Gene arrangement was highly conserved across Diptera. The relative synonymous codon usage (RSCU) varied among families (Figure S1).
Transcriptomes from eight species were assessed for completeness using BUSCO and assembly metrics (Figure 2, Table S2). Most species demonstrate reliable assembly statistics, with the number of assembled transcripts ranging from 25,847 in Blepharicera sp1.-QZ to 53,632 in Blephariceridae sp1.-WYM. The GC content varies from 29.86% in Deuterophlebia wuyishanense to 41.39% in Blephariceridae sp1.-WYM. The BUSCO assessments confirmed high assembly quality, with most species containing over 2000 single-copy orthologs, except for Blephariceridae sp1.-WYM (Figure 2, Table S2).
2.2. Phylogenetic Analyses Using Nuclear Datasets
Our phylogenetic reconstruction using nuclear genomic datasets with a varying number of loci and site occupancy levels for both amino acids and nucleotides consistently rejected the monophyly of Blephariceromorpha. Instead, all analyses strongly supported Deuterophlebiidae and Nymphomyiidae as forming the basal-most position of Diptera, while placing Blephariceridae within Psychodomorpha. The amino-acid-based phylogenies (Figure 3 and Figure 4) supported a well-resolved topology for Diptera, showing the following: (1) Culicomorpha diverged, representing the most basal lineage except Deuterophlebiidae + Nymphomyiidae; (2) a sister relationship between Tipulomorpha and Ptychopteromorpha; and (3) these successive clades collectively forming the sister group to the remaining Diptera. In contrast, nucleotide-based analyses produced a less robust alternative topology (bootstrap support <90% for key nodes), with (Tipulomorpha + Ptychopteromorpha) diverged first, followed by a clade comprising Culicomorpha sister to remaining dipteran groups.
We employed DiscoVista to explore potential phylogenetic discordance between amino acid and nucleotide datasets. The results strongly supported most major dipteran clades but rejected the monophyly of Blephariceromorpha (Figure 4). Specifically, when Blephariceridae was grouped within Psychodomorpha, and Deuterophlebiidae, and Nymphomyiidae were positioned as the basal-most lineage, the phylogenetic trees received robust support. The comparative analysis revealed amino acid trees consistently recovered established dipteran suborders and most infraorders. In contrast, nucleotide trees failed to support monophyly for several groups including Culicomorpha, Bibionomorpha, and Psychodomorpha. This demonstrates that amino-acid-based analyses were more reliable in terms of node support and topological consistency. In addition, we further employed FcLM to examine the relationships among Blephariceridae, Deuterophlebiidae, and Nymphomyiidae within the broader context of dipteran groups (Figure 5). The nuclear gene datasets indicated a closer relationship between Deuterophlebiidae and Nymphomyiidae, while Blephariceridae appeared more closely related to other dipteran groups, particularly in the amino acid datasets.
Finally, we generated consensus trees from our amino acid and nucleotide datasets using DensiTree (v 3.1.0) (Figures S2 and S3). While the amino-acid-based and nucleotide-based trees exhibited distinct topologies, they both consistently identified the basal-most position of Diptera as a clade consisting of (Deuterophlebiidae + Nymphomyiidae). This consistency highlights the reliability of nuclear genomic data in resolving early branching events in Diptera evolution.
2.3. Phylogenetic Analyses Using Mitochondrial Datasets
Phylogenetic analyses were performed using mitochondrial genomes from 114 species, including our newly sequenced 11 Blephariceromorpha species. While the monophyly of each family (Blephariceridae, Deuterophlebiidae, and Nymphomyiidae) was strongly supported, relationships among higher taxa showed inconsistencies and low support (bootstrap support <80% for key nodes; Figure S4, Supplementary Files). Notably, the phylogenetic positions of Blephariceridae, Deuterophlebiidae, and Nymphomyiidae exhibited significant variations across the datasets.
To further assess the consistency of the phylogenetic trees, we used DiscoVista to test specific hypotheses concerning the monophyly of Blephariceromorpha and the potential placement of Deuterophlebiidae and Nymphomyiidae as basal lineages of Diptera (Figure S4). The analyses rejected monophyly for most traditionally defined suborders and infraorders of Diptera, including Blephariceromorpha, aligning with the weak nodal bootstrap support observed. Additionally, likelihood mapping analysis (Figure 5), which examined relationships between Blephariceridae, Deuterophlebiidae, Nymphomyiidae, and other dipteran taxa, revealed a topology where Blephariceridae and Deuterophlebiidae formed a group, while Nymphomyiidae appeared more closely related to the remaining Diptera.
Attempts to visualize the mitochondrial trees using DensiTree (v 3.1.0) to derive a consensus tree were unsuccessful. Consequently, the relationships among the three families within Blephariceromorpha and the basal lineages of Diptera remain unresolved, highlighting the persistent challenges requiring complementary nuclear genomic approaches to clarify these basal dipteran relationships.
3. Discussion
3.1. New Insights into Phylogeny of Nematocera
This study reconstructed dipteran phylogeny, with a particular focus on Nematocera, using a comprehensive dataset representing 64 families (>100 species) with Mecoptera and Siphonaptera as outgroups. Our transcriptome assembly quality was rigorously assessed using the diptera_odb10 BUSCO set (v5.6.1) [28], which currently provides the most comprehensive benchmark for dipteran phylogenomics (3285 genes from 56 species). For our focal taxa, RNA-Seq data yielded over 1500 high-confidence single-copy orthologs per species at 80% occupancy thresholds, collectively providing phylogenomic-grade data comparable to genome-derived markers.
The results demonstrated that nuclear genomic data yield superior phylogenetic inferences, with amino-acid-based trees consistently recovering monophyletic major clades (Figure 3). To explore these inconsistencies, we employed FcLM and DiscoVista (v1.0) to assess discordances across datasets and tree topologies. While mitochondrial genomes remain valuable for phylogenetic studies at shallow taxonomic levels due to their compact size, maternal inheritance, and elevated mutation rates [23,29,30], our analyses revealed significant limitations at deeper evolutionary timescales. Mitochondrial gene trees exhibited artifacts including long-branch attraction and substitution saturation [31,32,33,34], whereas nuclear genomic datasets demonstrated a superior phylogenetic performance, owing to their multi-locus nature and lower susceptibility to systematic biases. These findings corroborated an emerging consensus that genome-scale nuclear data provide more reliable phylogenetic signal for deep divergences [35,36,37]. Comparative analyses further showed that amino acid sequences outperformed nucleotide data by producing higher clade support values (FcLM/DiscoVista) and showing greater topological consistency. This aligns with the established understanding that amino acid substitutions are less affected by saturation and compositional heterogeneity, both of which are known to compromise phylogenetic accuracy [26,38,39,40].
By integrating these findings, we constructed a highly supported phylogenetic tree for Diptera, with a particular focus on the suborder Nematocera. The monophyly of each dipteran family was consistently reinforced, and relationships within the suborder showed variation depending on the dataset and method used. Earlier studies of Nematocera phylogeny, excluding Deuterophlebiidae and Nymphomyiidae, proposed a topology based on morphological evidence: (Culicomorpha + (Tipulomorpha + ((Blephariceridae + Tanyderidae) + ((Psychodidae + (Trichoceridae + Bibionomorpha)) + Brachycera)))) [2]. Another study, which combined morphological and molecular data, suggested the following widely accepted topology: (Tipulomorpha + (Psychodomorpha + (Culicomorpha + (Bibionomorpha + Brachycera)))) [18]. In addition, a comprehensive Insecta Phylogenomic study revealed an alternative topology in which Culicomorpha diverged earlier than Tipulomorpha [24].
In our study, which used similar methodologies to the Insecta Phylogenomics by employing large nuclear gene datasets for phylogenetic analysis of Diptera, we found that amino acid data supported the topology ((Deuterophlebiidae + Nymphomyiidae) + (Culicomorpha + ((Ptychopteromorpha + Tipulomorpha) + (Psychodomorpha + (Bibionomorpha + Brachycera))))), while nucleotide data produced results more consistent with classical dipteran phylogenies (Figure 3, Figure 4, Figures S3 and S4 Supplementary Files).
While chromosome-level genomes remain ideal, their current unavailability for Deuterophlebiidae, Nymphomyiidae, and Blephariceridae highlights the importance of our transcriptome-based approach. Despite significant progress in Diptera genomics, basal nematoceran lineages continue to represent a critical sampling gap in current genomic resources. To address this limitation, we recommend that future studies incorporate the newly released BUSCO sets (diptera_odb12 and nematocera_odb12) [41] with RNA-Seq data, as our results demonstrate that transcriptome sequencing provides a phylogenomic-grade resolution for non-model organisms [25]. Our comprehensive analyses demonstrate that nuclear gene-based phylogenetic trees exhibit remarkable consistency across analytical methods when using amino acid sequences, in contrast to the methodological sensitivity observed in nucleotide-based approaches [26,38,39,40]. As a result, we present a novel topology for Diptera, offering broader taxonomic and gene representation compared to previous studies.
3.2. Investigation of Basal Lineages in Diptera
Our research placed particular emphasis on elucidating the phylogenetic positions of basal dipteran lineages, with special attention to the infraorder Blephariceromorpha. Traditional taxonomic classifications have proposed that Blephariceromorpha comprises Deuterophlebiidae, Nymphomyiidae, and Blephariceridae, based on several lines of evidence including shared ecological characteristics, overlapping geological distributions, and morphological features [13,15,42]. Nevertheless, these studies, often relying on morphological data or limited nuclear and mitochondrial gene datasets, have yielded inconsistent and debated conclusions regarding the positions of basal lineages and the monophyly of Blephariceromorpha.
Emerging phylogenetic studies have begun to clarify the positions of these enigmatic basal dipteran lineages, suggesting that Deuterophlebiidae represents a basal lineage of Diptera, while Blephariceridae is placed within Psychodomorpha [2,14]. However, the phylogenetic position of Nymphomyiidae remains unresolved. Previous morphological phylogenetic studies, limited by the lack of Deuterophlebiidae samples, supported Nymphomyiidae as a basal lineage of Diptera [18]. In addition, Wiegmann et al. proposed a sister-group relationship between (Deuterophlebiidae + (Nymphomyiidae + other Diptera species)) [2], while recent studies using mitochondrial genomes suggest that Nymphomyiidae belongs within Culicomorpha [14,20]. These conflicting results highlight the challenges in resolving the phylogenetic relationships of the basal dipteran lineages.
In our study, largely consistent with previous findings, the monophyly of Blephariceromorpha was rejected. We found that Blephariceridae is more closely aligned with Psychodomorpha, while Nymphomyiidae and Deuterophlebiidae form a sister clade, positioned as the basal lineage of Diptera. This conclusion is supported by morphological evidence, such as the loss of mouthparts, elongation of the terminal antennal segment, and the presence of sucker-baring larval prolegs adapted to fast-flowing streams in both Deuterophlebiidae and Nymphomyiidae, indicating a close evolutionary relationship between these two families [19].
Our study reconciles earlier morphological and molecular phylogenetic perspectives, offering a unified understanding of the evolutionary relationships within Diptera. By integrating comprehensive molecular datasets, including complete mitochondrial genomes and transcriptomic data, our research represents a significant advancement in dipteran phylogeny. This robust approach provides a more detailed and reliable phylogenetic analysis, overcoming the limitations of previous studies that relied on smaller, less informative datasets.
3.3. Convergent Evolution of Larval Morphology in Blephariceromorpha
The three families within the traditionally defined infraorder Blephariceromorpha—Blephariceridae, Deuterophlebiidae, and Nymphomyiidae—exhibit striking similarities in their larval morphological adaptations (e.g., prolegs) to fast-flowing stream environments. These shared ecological specializations, compounded with overlapping geological distribution, have historically justified their grouping. A central question in Dipteran phylogeny has been whether these similarities result from a single evolutionary origin or multiple instances of convergent evolution.
Our comprehensive molecular analysis provides decisive evidence on this question. Phylogenetic trees derived from nuclear datasets support the paraphyly of Blephariceromorpha, indicating that Deuterophlebiidae and Nymphomyiidae share a common ancestor with similar larval adaptations, while Blephariceridae likely evolved these traits independently. This pattern strongly suggests that the demanding elective pressures of fast-flowing stream environments have driven convergent evolution. Key adaptations like specialized prolegs for substrate attachment and streamlined body shapes appear to have emerged multiple times under similar ecological constraints. This supports broader theories of convergent evolution, where ecological niches often drive the repeated evolution of similar traits, even in distantly related species [43,44,45]. Notable examples include fang evolution in frogs [46], the springboard trapping mechanism in carnivorous pitcher plants [47], the transition from egg-laying to live-bearing in marine snails [48], and the compound eyes in the chitons [49]. In the case of Blephariceromorpha, the similar larval morphologies exemplify morphological and ecological convergence, specifically highlighting adaptations such as abdominal suckers and spinule-equipped prolegs. However, Blephariceridae larvae possess abdominal suckers combined with non-suckered prolegs, while those of Deuterophlebiidae and Nymphomyiidae exhibit spinule-equipped prolegs. This suggests that the origin of these structures, adapted for attachment in fast-flowing environments, reflects non-homologous but functionally analogous traits, highlighting their similar ecological roles despite different developmental origins.
A deeper understanding of the developmental mechanisms and gene regulatory networks underlying the convergent evolution of larval adaptations in fast-flowing environments is crucial to fully elucidating the evolutionary processes shaping these lineages. Investigating developmental pathways, such as gene regulation and morphogenetic patterns, could further reveal how different lineages independently evolved similar traits in response to parallel environmental pressures.
4. Materials and Methods
4.1. Sample Collection
Specimens of Diptera were collected from Yaowang Mountain, Zhejiang, China, and Wuyi Mountains, Fujian, China (Table 1). Fresh samples were preserved in RNAlater^®^ (Thermo Fisher Scientific, Waltham, MA, USA) for RNA extraction and in 70% ethanol for DNA extraction. The samples were stored at −80 °C (School of Life Sciences, Westlake University, Hangzhou, Zhejiang Province, China).
4.2. DNA Extraction, Mitogenomes Sequencing, and Analysis
The genomic DNA was extracted from undissected whole-body specimens of each species using the Qiagen DNeasy Blood and Tissue kit (Hilden, Germany), following the manufacturer’s protocol. Illumina Truseq libraries (San Diego, CA, USA) were prepared for each species, with an average insert size of 400 base pairs (bp). These libraries were then sequenced on the Illumina NovaSeq 6000 platform, generating 150 bp paired-end reads.
Adaptor sequences and low-quality reads were trimmed with Trimmomatic (v0.39) [50]. The mitochondrial genome of each species was assembled and annotated using Getorganelle (v1.7.7) [51], MITOS2 (v2.1.0) [52] and Mitofinder (v1.4.1) [53].
4.3. RNA Extraction, Sequencing, and Transcriptome Assembly
Total RNA was extracted from RNAlater-preserved samples using TRIzol (Thermo Fisher Scientific, Waltham, MA, USA). RNA sequencing libraries were prepared using the NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA), following the manufacturer’s instructions. RNA-seq libraries were sequenced on the Illumina HiSeq 2000 platform, generating 150 bp paired-end reads.
Raw sequencing reads were quality-trimmed using Trimmomatic (v0.39) [50], and transcriptome assembly was performed with Trinity (v2.15.1) [54]. The longest isoform per locus was extracted. The transcriptome assembly quality was assessed with BUSCO v5.6.1 (--mode transcriptome) [28], using the Diptera-specific ortholog set (diptera_odb10). This benchmark dataset comprises 3285 single-copy BUSCO genes conserved in 56 dipteran species (Eukaryota domain), with maximum intron length (≤130 kb) and sequence length (≤160 kb) thresholds.
4.4. Matrix Construction
Phylogenetic trees were reconstructed based on two datasets: mitochondrial genomes (114 species) [14,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102] and nuclear genes (126 species) [103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135], both including 61 families of Diptera (Tables S3 and S4). The outgroup taxa (Mecoptera and Siphonaptera) were selected based on their well-established sister-group relationship with Diptera in molecular phylogenies [2,24], providing appropriate phylogenetic distance while minimizing long-branch artifacts.
For mitogenomes, the data analysis was conducted in PhyloSuite (v1.2.3) [136]. Sequences of protein-coding genes (PCGs) and RNA genes were aligned individually via the MAFFT (v7.520) [137]. The alignments were manually reviewed in MEGA (7.0) [138], and poorly aligned regions were removed using GBLOCKS (v.0.91b) [139]. The individual gene alignments were then concatenated with SEQUENCEMATRIX (v.1.8) [140] to generate the following three datasets: (1) 13PCGs_NT12 matrix, including the first and second codon positions of 13 PCGs (7360 sites); (2) 13PCGs_NT123 matrix, including all three codon positions of 13 PCGs (total of 11,040 sites); and (3) 13PCGs_AA matrix, including the amino acids of 13 PCGs (3555 sites).
For the nuclear genes, universal single-copy ortholog (USCO) sequences were extracted following the workflow “script2_BUSCO_extraction.sh” in PLWS (v1.0.6) [141]. Briefly, coding regions were first identified from nucleotide sequences and translated into protein sequences using TransDecoder (v5.5.0) [142]. Protein sequences were then aligned with MAFFT (v7.520) [137], and low-quality or highly variable regions were trimmed using TrimAl (v1.4.1) [143]. Finally, the refined protein alignments were concatenated into a single dataset using FASconCAT-G (v1.05.1) [144]. Five phylogenetic matrices were generated with strict per-locus species coverage thresholds (50%, 60%, 70%, 80%, and 90%), where each retained locus contained sequences from ≥the specified percentage of species. Separate nucleotide and amino acid datasets were constructed for each threshold (Table 2).
4.5. Phylogenetic Analysis
Phylogenetic analyses of the mitogenome datasets were conducted using Bayesian inference (BI), maximum likelihood (ML), and ASTRAL-III methods. For the nuclear gene datasets, we employed ML and ASTRAL-III for tree construction.
Maximum Likelihood (ML) analyses were performed using IQ-TREE (v2.3.4) [145]. ModelFinder (-m MFP --mset GTR, LG) [146] was utilized to select the optimal partitioning scheme and substitution models for both nucleotide and amino acid data (GTR for nucleotides, LG for amino acids). Alternatively, the GHOST model (-m GTR+FO+H4, LG+FO+H4) [147] was applied to address heterotachy (site-specific evolutionary rate variation across branches). Phylogenetic reconstructions were performed with ultrafast bootstrap approximation (UFBoot) [148] using 1000 replicates (-B 1000). Branch support was further strengthened by incorporating the SH-aLRT test (-alrt 1000) [149] and refining the trees through branch nearest-neighbor interchange (--bnni). Additionally, the rcluster algorithm (-rcluster 10) [150] with a threshold of 10 was employed to enhance both tree accuracy and efficiency.
Individual gene trees were inferred with IQ-TREE with automatically selected best models and analyzed with ASTRAL-III (v5.6.1) [151] to infer the coalescent-based species trees, with local branch supports estimated from quartet frequencies.
Bayesian inference (BI) was conducted using PhyloBayes MPI (v.1.9) [152] under the site-heterogeneous mixture models CAT + GTR or CAT + LG, with a discrete gamma distribution and four rate categories for nucleotide or amino acid datasets. Two independent Markov Chain Monte Carlo (MCMC) chains were run until convergence was achieved (maxdiff < 0.1). After discarding the first 25% of trees as burn-in, a consensus tree was computed from the remaining trees combined from both runs.
Tree visualization and modifications were performed using the ITOL web server (v6) [153], Figtree (v1.4.4) [154], ggtree (v3.10.0) [155], and Adobe Illustrator (v2022).
4.6. Analyses of Phylogenetic Discordance and Alternative Relationships
DiscoVista (v1.0) [156] was used to quantify and visualize discordance among phylogenetic trees for alternative topologies. Our analysis primarily focused on the monophyly of Diptera groups, with particular emphasis on Blephariceromorpha, using a bootstrap threshold of 95 (-t 95).
To further investigate potential incongruence or confounding signals within the nuclear and mitochondrial gene datasets, especially regarding the relationships among Blephariceridae, Deuterophlebiidae, and Nymphomyiidae, we applied four-cluster likelihood mapping (FcLM) [157] implemented in IQ-TREE (-lmap, -m GTR for nucleotides, LG for amino acids) [145] to display support for the three possible quartet topologies and visualize the strength of each alternative relationship.
5. Conclusions
Overall, this study provides a comprehensive analysis of the phylogenetic relationships within Diptera, particularly the basal lineages, utilizing a combination of mitochondrial and nuclear gene datasets. While some topological inconsistencies emerged between different analytical approaches and data types, several key findings received robust support. The majority of our phylogenetic reconstructions decisively rejected the monophyly of Blephariceromorpha. Notably, the findings establish Deuterophlebiidae + Nymphomyiidae as the basal-most lineage of and the sister group to the rest of Diptera, whereas Blephariceridae was found to be grouped within the Psychodomorpha, a clade distantly related to Deuterophlebiidae and Nymphomyiidae. This novel phylogenetic arrangement suggests convergent evolution as the underlying mechanism for the remarkably similar larval adaptations observed in these two groups, Blephariceridae and (Deuterophlebiidae + Nymphomyiidae). These insights contribute to our understanding of the evolutionary history and diversification of Diptera, highlighting the importance of comprehensive molecular datasets and rigorous phylogenetic analyses in resolving complex evolutionary relationships.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Systema Dipterorum Available online: http://www.diptera.org/(accessed on 5 September 2024)
- 2Wiegmann B.M. Trautwein M.D. Winkler I.S. Barr N.B. Kim J.-W. Lambkin C. Bertone M.A. Cassel B.K. Bayless K.M. Heimberg A.M. Episodic Radiations in the Fly Tree of Life Proc. Natl. Acad. Sci. USA 20111085690569510.1073/pnas.101267510821402926 PMC 3078341 · doi ↗ · pubmed ↗
- 3Yeates D. Wiegmann B. Phylogeny and Evolution of Diptera: Recent Insights and New Perspectivs The Evolutionary Biology of Flies Columbia University Press New York, NY, USA 2005
- 4Yeates D.K. Wiegmann B.M. Courtney G.W. Meier R. Lambkin C. Pape T. Phylogeny and Systematics of Diptera: Two Decades of Progress and Prospects Zootaxa 2007166856559010.11646/zootaxa.1668.1.27 · doi ↗
- 5Yeates D. Wiegmann B. Phylogeny of Diptera Manual of Afrotropical Diptera SANBI Graphics & Editing Pretoria, South Africa 2017 Volume 4253265
- 6Borkent A. The State of Phylogenetic Analysis: Narrow Visions and Simple Answers-Examples from the Diptera (Flies)Zootaxa 2018437410714310.11646/zootaxa.4374.1.729689817 · doi ↗ · pubmed ↗
- 7Savage J. Borkent A. Brodo F. Cumming J.M. Curler G. Currie D.C. de Waard J.R. Gibson J.F. Hauser M. Laplante L. Diptera of Canada Zoo Keys 201981939745010.3897/zookeys.819.2762530713456 PMC 6355757 · doi ↗ · pubmed ↗
- 8Gerhardt R.R. Hribar L.J. Chapter 11—Flies (Diptera). In Medical and Veterinary Entomology 3rd ed. Mullen G.R. Durden L.A. Academic Press Cambridge, MA, USA 2019171190978-0-12-814043-7