Variant Ataxia–Telangiectasia Presenting as Tremor–Dystonia Syndrome in a Bulgarian Religious Minority
Teodora Chamova, Tihomir Todorov, Paulius Palaima, Petya Yankova, Iliyana Pacheva, Ivan Ivanov, Bilyana Georgieva, Sylvia Cherninkova, Alexey Savov, Dora Zlatareva, Elisaveta Naumova, Albena Todorova, Albena Jordanova, Ivailo Tournev

TL;DR
A rare genetic disorder caused by ATM gene mutations can present as tremor and dystonia without typical brain symptoms, highlighting the need for broader diagnostic consideration.
Contribution
Identifies a novel variant form of Ataxia-telangiectasia with dystonia and tremor as primary symptoms, not previously well characterized.
Findings
Patients with ATM gene mutations presented dystonia and tremor without significant cerebellar involvement.
Most patients were homozygous for the c.8147T>C mutation in ATM, with some compound heterozygous mutations.
Clinical onset varied widely, from infancy to adulthood, with slowly progressive symptoms and normal brain imaging in most.
Abstract
Background: Ataxia-telangiectasia (A-T) is a rare autosomal recessive disorder due to mutations in the ATM gene. Given the residual kinase activity and the type of ATM mutation, its clinical spectrum varies from a severe classic phenotype to a variant atypical form. Material and methods: This study included 28 patients belonging to four big Bulgarian Muslim pedigrees with tremor and dystonia. Whole-exome sequencing was performed in seven affected individuals from two unrelated pedigrees, followed by Sanger sequencing of the coding sequences and exon–intron borders of the ATM gene. Results: Twenty-four of the affected individuals were homozygous for c.8147T>C (p.Val2716Ala) in ATM, while four of the affected individuals were compound heterozygous. The targeted Sanger sequencing along the ATM gene revealed as a second mutation in three of the patients the splice-site variant c.4909+1G>A…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
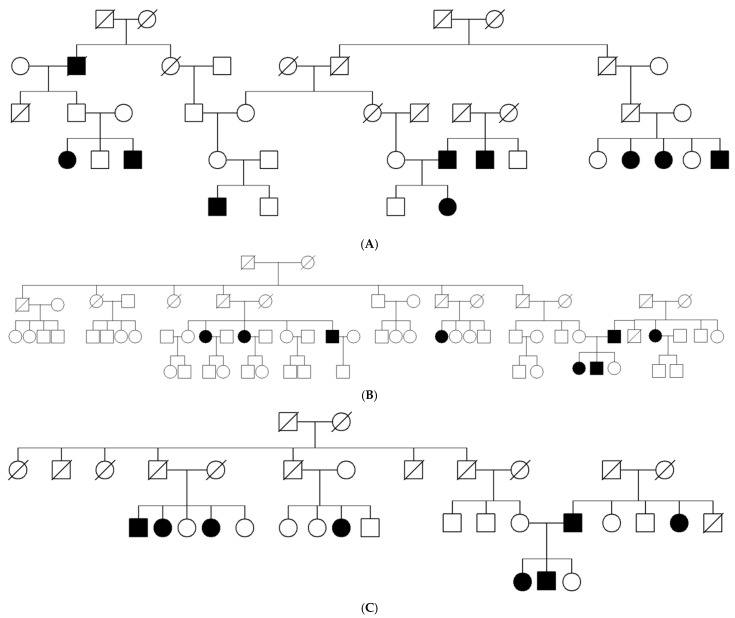 Figure 1
Figure 1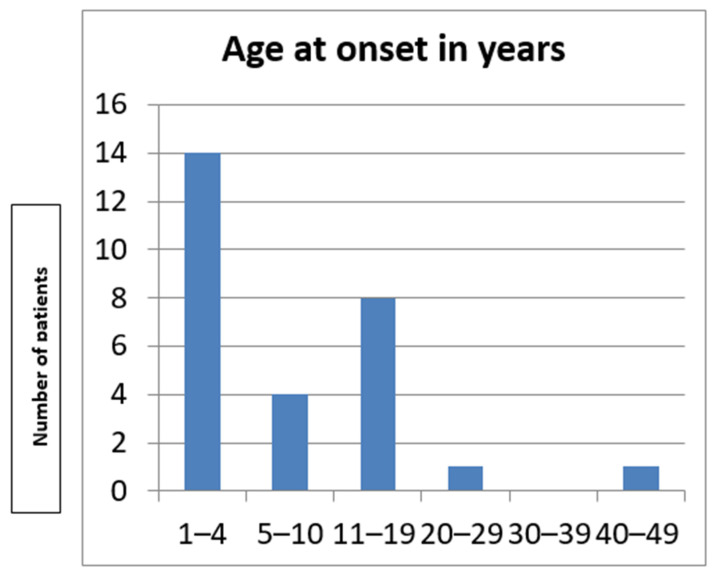 Figure 2
Figure 2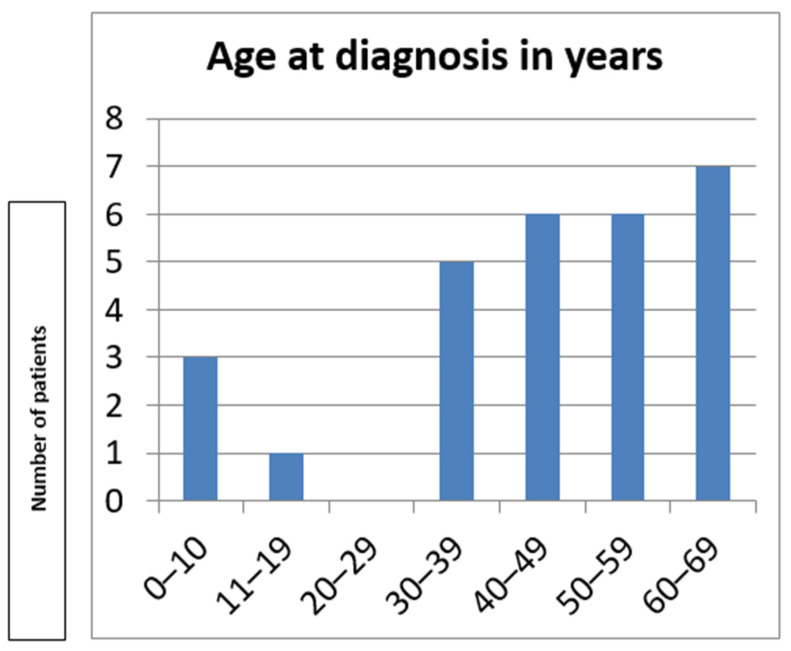 Figure 3
Figure 3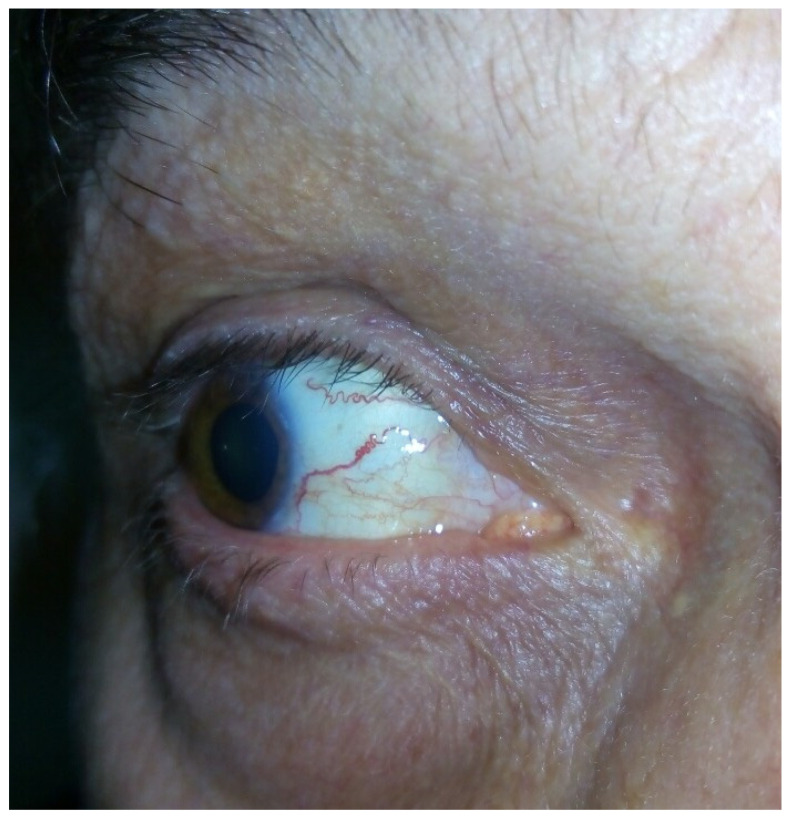 Figure 4
Figure 4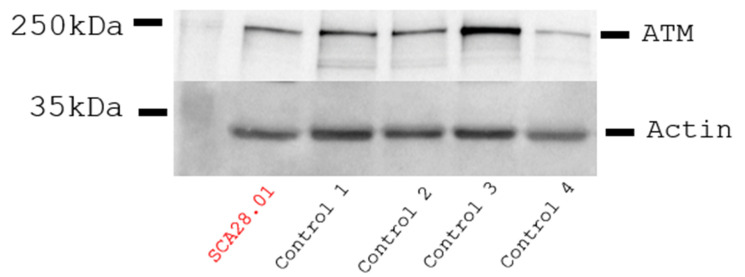 Figure 5
Figure 5- —National Recovery and Resilience Plan of the Republic of Bulgaria
- —Fund for Scientific Research (FWO-Flanders)
- —French Muscular Dystrophy Association
- —Research Fund of Medical University-Sofia
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic Neurodegenerative Diseases · DNA Repair Mechanisms · Neurological disorders and treatments
1. Introduction
Ataxia–telangiectasia is a rare autosomal recessive disorder caused by mutations in the ATM gene, located on chromosome 11q22.3 (MIM 208900) [1]. This gene is known to harbor 66 exons and encodes a PI3K family kinase with 3050 amino acid residues. Currently, more than 1400 unique variants have been identified in this gene [2]. The ataxia–telangiectasia-mutated (ATM) protein is a serine-threonine protein kinase, which phosphorylates more than 700 substrates and is a key player in the cellular response to double-stranded DNA damage [3]. Due to the residual kinase activity and the nature of the ATM mutation, the clinical presentation of AT can vary widely—from the classic form characterized by severe, early-onset, and rapidly progressive neurodegeneration to an atypical variant with milder neurological symptoms and a reduced risk of systemic complications. Patients with classic ataxia–telangiectasia present with a severe phenotype due to absent ATM kinase activity [4], either attributed to two null mutations or mutations that result in protein without ATM kinase activity. Affected individuals have severe multisystem involvement, including cerebellar ataxia, tremor, dystonia, oculomotor dyspraxia, polyneuropathy, immunological defects, respiratory problems, oculocutaneous telangiectasia, and an increased risk of malignancy [5,6,7]. Most patients with classic ataxia–telangiectasia lose ambulation during adolescence and die before the age of 30 due to malignancy or respiratory failure.
The term ‘‘variant A-T’’ is used to describe patients with milder phenotypes, associated with missense or splice-site mutations that produce ATM proteins with some residual kinase activity [8]. The variant A-T patients have a milder late-onset atypical phenotype characterized by a predominance of tremor, dystonia, ataxia with later onset, and slower neurologic progression. Although respiratory diseases and immunodeficiency are less severe in variant A-T, the risk of malignancy remains high [4,6,9].
Here, we report the clinical features of a cohort of 28 patients with variant ataxia–telangiectasia caused by the missense c.8147T>C, p.Val2716Ala and the splice-site c.4909+1G>A and c.3576G>A, p.Lys1192 variants and who belong to a religious minority from the southwest of Bulgaria.
2. Materials and Methods
This study encompassed 28 patients (12 male and 16 female) from four unrelated pedigrees belonging to a religious minority in Bulgaria. The families were identified through field studies and records from the Department of Neurology at the Medical University of Sofia. All participants provided written informed consent. This study adheres to the ethical guidelines of the participating institutions. It was approved by the Ethics Committee of Sofia Medical University, 4873, approved on 9 July 2018.
All the affected patients underwent clinical neurological assessment, including the Scale for Assessment and Rating of Ataxia (SARA) and Unified Dystonia Rating Scale (UDRS) [10,11]. According to the predominant neurological symptoms, the patients were divided into three neurological phenotypic groups: Group 1, cerebellar ataxia with minimal or no extrapyramidal involvement; Group 2, cerebellar ataxia combined with additional extrapyramidal features; and Group 3, extrapyramidal signs without notable ataxia [12].
Each patient’s medical history, including any instances of diabetes and malignancy, was thoroughly documented.
Height and weight were collected from all of the affected individuals and compared to the normative values of the Bulgarian population.
Neuro-ophthalmological assessment for conjunctival telangiectasia and eye movement abnormalities was performed in all of the affected patients. Nerve conduction studies were conducted in 16 of the affected individuals.
MRI brain scans were conducted by a 3 Tesla MRI unit (Siemens Verio) in 14 patients from this cohort. Brain CT scans were conducted in 3 affected patients. Alpha fetoprotein in blood (AFP) was tested in 14 patients.
2.1. Immunological Assessment
History of infections was assessed in all patients. Immunological examination of four patients was performed, including the immunophenotyping of lymphocytes (T lymphocytes and their subpopulations: Ts, Th, Th17, and Treg; B and NK cells), T cell function during stimulation with PHA and anti-CD3/CD28 dynabeads, determination of immunoglobulins (IgG and their subclasses,: IgA, IgM, and IgE), complement fractions (C3 and C4), antinuclear antibodies (ANA), and post-vaccinal immune response against tetanus and diphtheria anatoxin. Indirect immunofluorescence (IIF) with HEp-2 cells was used as a screening technique for the detection of antinuclear autoantibodies. Flow cytometry analyses were used to detect lymphocyte populations and functional markers on T cells [13].
2.2. Whole-Exome Sequencing and Homozygosity Mapping
Genomic DNA was isolated from peripheral blood mononuclear cells following standard procedures. Whole-exome sequencing (WES) was performed in two affected individuals and their parents from Family 1 from the town of Sarnitza and two affected siblings and their mother from Family 2 from the town of Dospat (Figure 1). Exome capture and enrichment was conducted using a SureSelect Human All Exon v5.0 kit (Agilent Technologies, Santa Clara, CA, USA), followed by paired-end sequencing on the HiSeq4000 platform (Illumina, San Diego, CA, USA). Primary analyses were conducted using the GenomeComb pipeline [14], which employed the Burrows–Wheeler algorithm [15] for read alignment to the GRCh37/HG19 reference human genome and genomic analysis toolkit (GATK) (version 3.3.0)22 for variant calling. Homozygosity mapping based on WES data was performed as described in [16] using the HOMWES tool from the GenomeComb package (version 0.11.0).20. Variant filtering and prioritization were performed using GenomeComb20 based on a recessive model of inheritance and the following criteria: variants within homozygous regions, non-synonymous or splice variants with a minor allele frequency (MAF) below 5%, and no homozygotes in the control population database gnomAD v2.1.1. [17]. Prioritized variants were evaluated further using Alamut Visual Plus (Sophia Genetics, Rolle, Switzerland).
2.3. Variant Segregation and Cohort Carrier Screening
The resulting variants were validated and segregated among the available family members using Sanger sequencing, as previously outlined.
Affected family members who turned out to be heterozygous for the variant c.8147T>C (p.Val2716Ala) were subjected to direct Sanger sequencing of the coding sequences and exon–intron borders of the ATM gene. The targeted screening along the ATM gene revealed two additional pathogenic variants, c.4909+1G>A and c.3576G>A, p.Lys1192, both of them affecting the normal splicing.
2.4. Screening in the Towns of Dospat and Surnica
Due to the fact that all patients belonged to a religious minority of Bulgarian Muslims originating from the Dospat and Surnica towns in the Rhodope Mountains, a selective screening targeting the most common mutation missense mutation c.8147T>C (p.Val2716Ala) was performed on newborns from each of both the presumable endemic regions. Altogether, 200 samples (100 for each region) were selected from a Guthrie card dry blood spot collection of national newborn screening programs (kindly provided by the National Genetic Laboratory, Sofia, Bulgaria). The study was approved by the Local Ethical Committee.
The screening for the targeted mutation was performed by PCR amplification followed by Sanger sequencing by a BigDye^®^Terminator cycle sequencing kit v.3.1 (Applied Biosystems, Foster City, CA, USA) [18]. Primers were designed to specifically amplify the targeted exon 57 of the ATM gene (RefSeq NM_000051). The electrophoretic separation was performed on an ABI3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The sequencing profiles were interpreted by the software Sequencing Analysis v5.1.1.
2.5. ATM Protein Expression
Total protein extracts were isolated from lymphoblast cells, separated on a custom-made 7.5% SDS-PAGE and transferred to blotting membranes as described in [19]. Membranes were incubated with the following primary antibodies: anti-ATM (Abcam, Cambridge, UK, 2C1 (1A1), 1:2000) and anti-actin (Merck, Darmstadt, Germany, A5441, 1:5000). Anti-mouse IgG1 (Southern Biotech, Birmingham, AL, USA, 1070-05, 1090-05, 1:10,000) was used as a secondary antibody. The blots were developed with Pierce ECL Plus substrate (Thermo Fisher Scientific, Waltham, MS, USA) and imaged using the Amersham Imager 600 (GE Healthcare, Chicago, IL, USA).
3. Results
The study included 28 individuals (12 male and 16 female) from four unrelated pedigrees (Figure 1) belonging to a religious minority of Bulgarian Muslims inhabiting the region of the Dospat and Surnica towns in the Rhodope Mountains. The mean age was 8.3 years ± 9.3 years, varying between 14 days and 40 years (Figure 2).
Mean age at diagnosis was 44.75 ± 18.03 years, ranging from 8 to 69 years (Figure 3). The diagnostic delay ranged between 5 and 58 years after the initial symptoms. None of the patients were wheelchair-bound at the time of the last assessment. No growth retardation was present in our cohort.
The main clinical features present in all of the affected individuals (Table 1) encompassed dystonia, resting, and postural tremor, predominantly affecting the head and the upper limbs (Supplementary Material). Dystonic dysarthria and dysphagia were observed in the more advanced stages. Chorea was observed in 10/28 of the patients, while myoclonus was present in 5/28. The movement disorders were slowly progressing over time. Mild ataxia of stance and gait was present only in 5/28 of the patients. All the affected individuals were ambulant at the time of their last assessment. In 4/28 patients, dilated conjunctival vessels were found (Figure 4).
Axonal polyneuropathy with decreased amplitudes of the peroneal and sural nerves was present in a small proportion of the patients: 4/17.
All the patients were classified in Group 3: extrapyramidal signs without notable ataxia and/or peripheral neuropathy. None of them were wheelchair-bound at the time of the last follow-up.
Brain MRI revealed cerebellar atrophy in only 1/17 of those tested (patient 28), who was compound heterozygous for p.V2716A and p.Lys119 in the ATM gene. Basal ganglia seemed unaffected as well.
Alpha fetoprotein was significantly increased in all of the 14 tested individuals, more than four times over the upper limit. None of the patients had a history of malignancy.
None of the patients reported severe infections during childhood. Although T cells were significantly diminished compared to standard values in three patients, the T cell subpopulations had a normal ratio. Only one patient (patient 28) showed severely reduced CD8+ T lymphocyte values. B cell lymphopenia and lower lymphocyte counts of regulatory T cells and elevated Th17 cell values were reported in all four patients. The immunological tests revealed normal expression of CD69+ total T lymphocytes with stimulation with PHA and anti-CD3/CD28 dynabeads and normal immunoglobulin and complement values. Non-protective post-vaccinal immune response against tetanus and diphtheria anatoxin and the titer of ANA was between 1/320 and 1/640.
Initial whole-exome sequencing followed by homozygosity mapping was conducted in seven patients and their available parents, belonging to three multigenerational families. The HOMWES analysis distinguished six regions of homozygosity shared between all of the affected individuals, with the largest one (9.2 Mb) located on chromosome 11 (chr11:102098354-111325051). Variant filtering and prioritization within those regions revealed a homozygous missense variant NM_000051.4(ATM):c.8147T>C (p.Val2716Ala) in exon 57 of the ATM gene. The variant was confirmed by Sanger sequencing, and the segregation analysis of available relatives demonstrated that it co-segregated with the disease. This variant was previously associated with variant A-T (ref). The immunoblotting analysis of total protein lysates isolated from lymphoblastoid cells available from patient 5 (SCA28.01) revealed that the AT gene was expressed at comparable levels to the gender- and age-matched control individuals (Figure 5).
Twenty-four of the affected patients were homozygous for c.8147T>C (p.Val2716Ala) in ATM, while four of the affected were compound heterozygous. The targeted Sanger sequencing along the ATM gene revealed as a second mutation in three of them the splice-site variant c.4909+1G>A and in one patient a synonymous pathogenic variant with a splicing effect, c.3576G>A, p.Lys1192.
Since there were cases in two consecutive generations (Figure 1) without a history of endogamy, the hypothesis of a high carrier rate of c.8147T>C (p.Val2716Ala) in ATM gene was tested. Altogether, 200 newborns originating from Dospat and Surnica were tested for the targeted mutation, c.8147T>C (p.Val2716Ala). The results showed three heterozygous carriers, which represents a carrier frequency in this subpopulation of about 1.5% (3/200). The mutant allele frequency was calculated on the base of 400 alleles screened, which revealed 0.75% (3/400).
4. Discussion
We present a large cohort of patients with a milder clinical phenotype, known as the A-T variant, belonging to a religious minority of Bulgarian Muslims. All individuals included in this study had neurological involvement, while systemic features were either mild or absent, which is in line with previous reports [12,20,21].
Despite genetic homogeneity in the ATM gene variants within our group, the age of onset varied widely, ranging from 14 days to 40 years. Such variation was observed in a group of Canadian Mennonites [20]. Consistent with other reports, tremor and dystonia, especially with facial, cervical, and brachial distribution, were invariably present in all the patients from the Bulgarian cohort [12,20], followed by chorea and myoclonus, observed in less than 50% of the affected patients in our group. They may occur without frank ataxia and may be misdiagnosed in adults with primary-appearing dystonia or essential tremor [12,20]. Mild ataxia was present in only 5/28 of the affected patients and did not progress significantly over time compared to the other movement disorders [21]. Moeini Shad et al. reported that patients with predominantly dystonic symptoms typically exhibit later disease onset and a milder clinical course—a pattern also observed in the Bulgarian cohort. However, it remains uncertain whether dystonia is associated with a specific genotypic abnormality [21].
In imaging of variant atypical AT patients, cerebellar morphology is often normal [22], as in our group only one patient had cerebellar atrophy on MRI.
It was reported that individuals with a missense mutation, which causes the production of a mutant ATM protein with retained kinase activity, are approximately five times more likely to have a malignancy than individuals in the other genetic groups due to a possible gain-of-function mechanism [12,20]. On the contrary, in our cohort, no malignancies have been reported up to now, but they are being thoroughly followed up.
AFP was elevated in all of the tested individuals from our cohort and may be considered as a useful screening tool with high sensitivity for A-T.
Immunodeficiency, though highly variable, is a hallmark of 60%–80% of patients with classic AT [3]; however, it is infrequently seen in variant forms of the disease [6]. We performed FACS analyses of T, B, and NK T cells from peripheral blood of four patients with the A-T variant to examine the expression of activation and functional markers. We observed a decrease in naïve helper (CD4+CD45RA+) cells, CD8+ T cells, and B cells and increased levels of Th17 cells, while the amount of CD3-CD56+ NK cells and CD3+CD56+ NKT-like cells was within the reference values corresponding to age. Despite the small number of patients studied, the results obtained are comparable to the results obtained by Graafen et al. [23]. In reviewing the available literature on elevated ANA values and their association with paraneoplastic syndrome in patients with the A-T variant, we found several findings that may confirm our results.
The most common mutation among this group, observed in a homozygous state in 24 affected patients, is p.Val2716Ala. The majority of Bulgarians are traditionally Christian, with only about 2% of the ethnic Bulgarian population belonging to the Muslim minority. This group, primarily concentrated in the Rhodope Mountains in the southwestern region of the country, was estimated at approximately 29,000 people according to the latest population census. This is the second genetic disorder after LGMD2G/R7 with a high frequency among this group [24,25]. In this region, the carrier rate of p.Val2716Ala was found to be quite high at 1.5% (3/200), probably due to cumulative effects of historical endogamy in this closed religious minority [24]. Our results indicate an expected high recurrence risk of variant ataxia–telangiectasia in these particular Bulgarian regions, which presumes the disease might be much more frequent than expected for the general population. These findings are important for the Muslim communities in the affected regions of Bulgaria. They could benefit from early genetic testing and appropriate genetic counseling during family planning.
5. Conclusions
Although there is no universally accepted definition of the variant AT phenotype, our findings align with previous reports indicating that variant A-T is characterized by tremor and dystonia, with or without overt ataxia. Because these cases are often underdiagnosed or misdiagnosed, early diagnosis through newborn screening and targeted testing in high-risk populations in Bulgaria is essential. This is particularly important given the associated risks of malignancies, as well as for facilitating genetic counseling, carrier detection within affected families, and prenatal diagnosis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Gatti R.A. Berkel I. Boder E. Braedt G. Charmley P. Concannon P. Ersoy F. Foroud T. Jaspers N.G. Lange K. Localization of an ataxiatelangiectasia gene to chromosome 11q 22-23Nature 198833657758010.1038/336577 a 03200306 · doi ↗ · pubmed ↗
- 2Shao L. Wang H. Xu J. Qi M. Yu Z. Zhang J. Ataxia-telangiectasia in China: A case report of a novel ATM variant and literature review Front. Neurol.202314122881010.3389/fneur.2023.122881037564729 PMC 10411728 · doi ↗ · pubmed ↗
- 3Mc Kinnon P.J. ATM and the molecular pathogenesis of ataxia telangiectasia Annu. Rev. Pathol.2012730332110.1146/annurev-pathol-011811-13250922035194 · doi ↗ · pubmed ↗
- 4Verhagen M.M. Last J.I. Hogervorst F.B. Smeets D.F. Roeleveld N. Verheijen F. Catsman-Berrevoets C.E. Wulffraat N.M. Cobben J.M. Hiel J. Presence of ATM protein and residual kinase activity correlates with the phenotype in ataxia-telangiectasia: A genotype-phenotype study Hum. Mutat.20123356157110.1002/humu.2201622213089 · doi ↗ · pubmed ↗
- 5Sedgwick R.P. Boder E. Ataxia telangiectasia Handbook of Clinical Neurology, Hereditary Neuropathies and Spino-Cerebellar Atrophies Vinken P.J. Bruyn S.W. Elsevier Science New York, NY, USA 1991347393
- 6Verhagen M.M. Abdo W.F. Willemsen M.A. Hogervorst F.B. Smeets D.F. Hiel J.A. Brunt E.R. van Rijn M.A. Majoor Krakauer D. Oldenburg R.A. Clinical spectrum of ataxia-telangiectasia in adulthood Neurology 20097343043710.1212/WNL.0b 013e 3181 af 33bd 19535770 · doi ↗ · pubmed ↗
- 7Woods C.G. Taylor A.M. Ataxia telangiectasia in the British Isles: The clinical and laboratory features of 70 affected individuals QJM Int. J. Med.1992821691791377828 · pubmed ↗
- 8Pearson T.S. More Than Ataxia: Hyperkinetic Movement Disorders in Childhood Autosomal Recessive Ataxia Syndromes Tremor. Other Hyperkinet. Mov.2016636810.5334/tohm.319PMC 495022327536460 · doi ↗ · pubmed ↗