The Integrative Taxonomy and Mitochondrial Genome Evolution of Freshwater Planarians (Platyhelminthes: Tricladida): The Discovery of a New Clade in Southern China
Yimeng Yang, Zhizhuo Huang, Xiaowen Fang, Pinyi Li, Yexin Li, Xiuying Hou, Yongjun Li, Hengwen Yang, Chunxia Jing, Zhinan Yin, Guang Yang

TL;DR
A new asexual freshwater flatworm species, Dugesia cantonensis, was discovered in southern China and shown to have strong regenerative abilities and a unique mitochondrial genome.
Contribution
The discovery of a new asexual Dugesia clade with a unique mitogenome and stable regeneration over multiple generations.
Findings
Dugesia cantonensis forms a distinct clade within Dugesia based on molecular and morphological data.
The species has a 18,125 bp mitogenome with 36 genes but lacks atp8.
All body fragments of D. cantonensis regenerate into complete individuals within nine days.
Abstract
Background: The genus Dugesia (Platyhelminthes: Tricladida) includes a large diversity of free-living freshwater flatworms and is important for studies on regeneration and evolution. This study aims to describe a newly discovered asexual planarian species from southern China and explore its genetic characteristics and regenerative abilities. Methods: An integrative taxonomic analysis was conducted using morphology, karyology, histology, molecular phylogeny (18S, 28S, COI, mitogenome), and genome size estimation via flow cytometry. Regeneration was assessed by standardized amputations, and long-term asexual propagation was observed under laboratory conditions for three years. Results: Phylogenetic analyses using nuclear (18S, 28S rDNA) and mitochondrial (COI, mitogenome) markers confirmed that Dugesia cantonensis Guang Yang & Zhinan Yin, sp. nov. forms a distinct clade within Dugesia.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
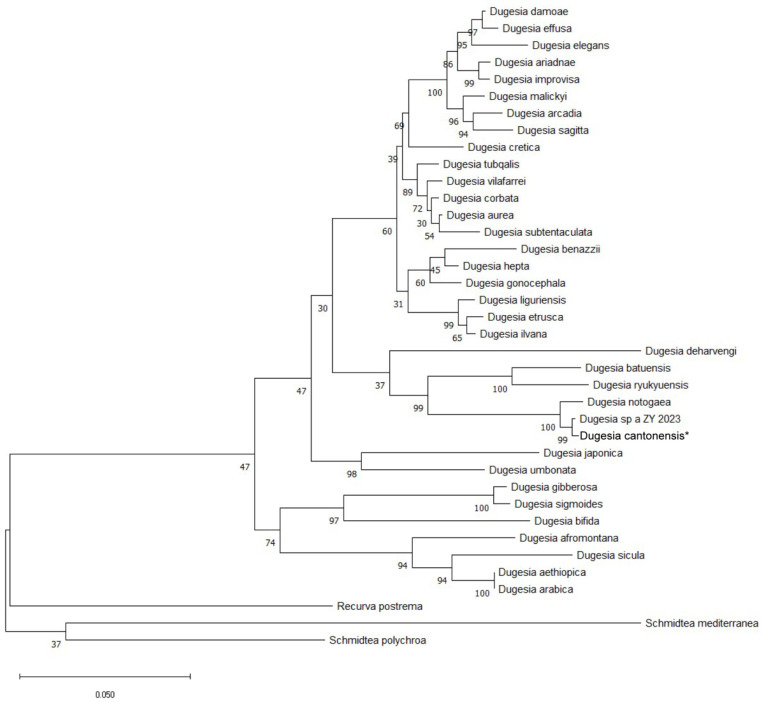 Figure 1
Figure 1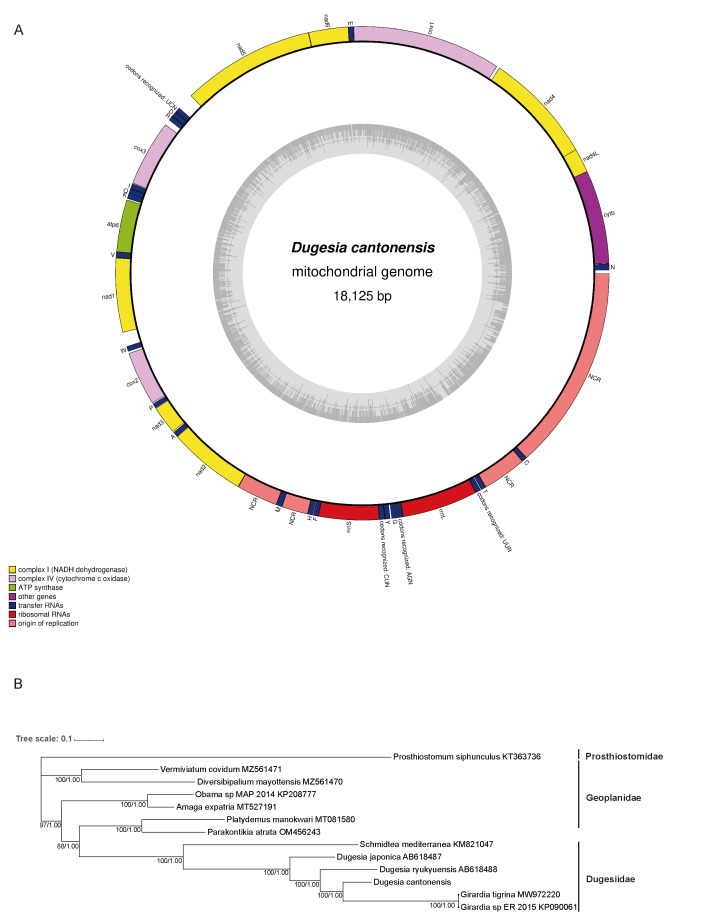 Figure 2
Figure 2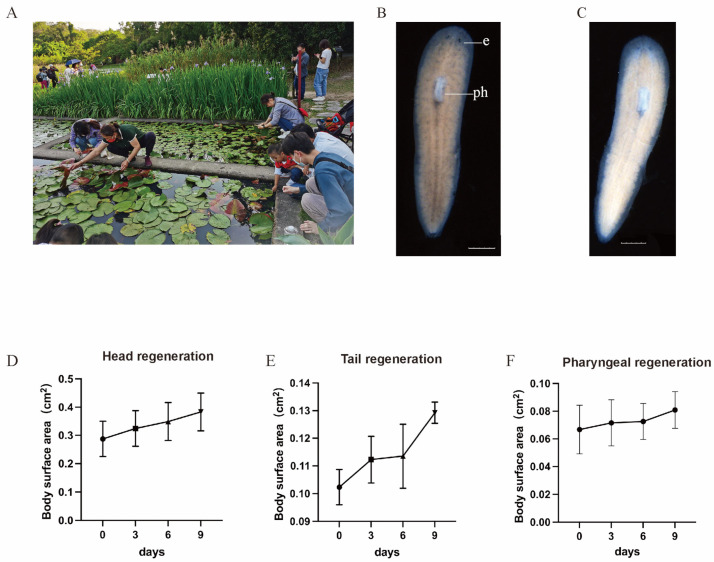 Figure 3
Figure 3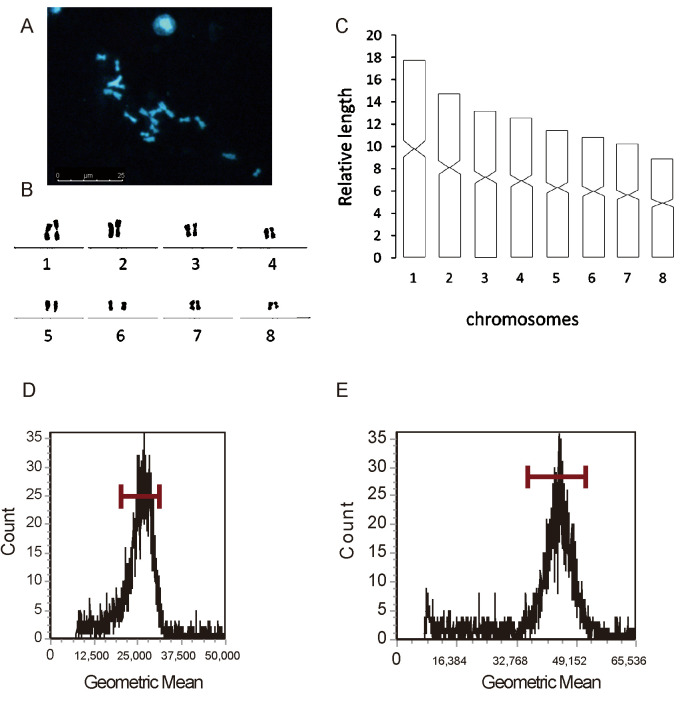 Figure 4
Figure 4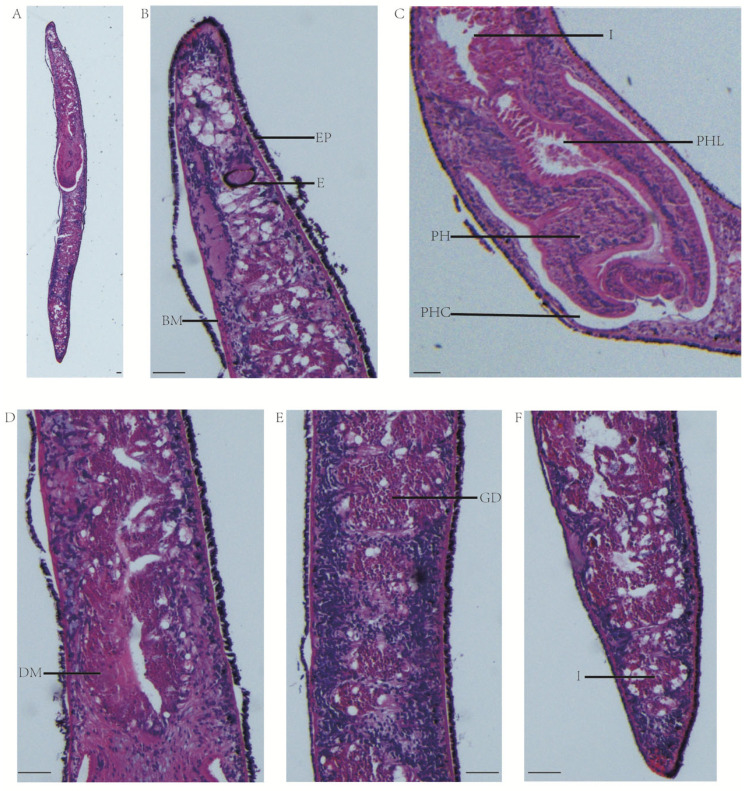 Figure 5
Figure 5- —Guangdong Basic and Applied Basic Research Foundation
- —Open Research Project of the Key Laboratory of Viral Pathogenesis & Infection Prevention and Control of the Ministry of Education
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlanarian Biology and Electrostimulation · Cephalopods and Marine Biology · Parasite Biology and Host Interactions
1. Introduction
The freshwater planarians of the genus Dugesia Girard, 1850, are the most primitive bilaterally symmetrical animals and occupy an important place in the history of species evolution [1]. Due to their undiminished regenerative ability, planarians have been chosen as one of the most appropriate experimental materials for stem cell research. About 100 species have been reported from most of the Old World and Australia [2]. However, for a long period after Dugesia japonica Ichikawa & Kawakatsu, 1964, was described from China, no new species of the genus were described, and it is only recently that taxonomic studies have begun to reveal the rich biodiversity of the genus in China [3,4,5,6]. Guangzhou is located in a subtropical coastal region, where the marine subtropical monsoon climate has fostered rich biodiversity. However, no Dugesiidae species have yet been recorded in this area. The discovery of a new species in Guangzhou would be an invaluable addition to the taxonomy of planarians in China. In this study, we aim to resolve the taxonomic challenges surrounding the evolutionary lineage and asexual strains of the Guangzhou planarian by establishing a localized strain and subsequently conducting investigations in regenerative biology, comparative genomics, and evolutionary biology.
Due to the great anatomical similarities between species, morphology-based phylogenetic analyses have struggled to resolve the affinities between species and species-groups [7]. The application of molecular markers offers a promising approach to overcome certain intrinsic constraints posed by morphological characteristics in species delineation and evolutionary history reconstruction among Dugesia [8]. Dugesia species exhibit a tripartite intestine, a T-shaped nervous system, and remarkable regenerative capabilities. Under suitable conditions, even small body fragments, including the head, tail, or trunk, can regenerate a fully functional organism.
The inclusion of karyological data may prove to be a highly informative additional line of evidence in systematic studies of the genus Dugesia. It has been reported that different Dugesia species exhibit differences in ploidy level and the centromeric position of the chromosomes [9,10,11]. Furthermore, several Dugesia species have been described with a chromosome portrait that differs from the most common haploid complement of n = 8, exhibiting complements such as n = 7 or n = 9 [12,13,14]. The results of chromosome karyotyping provide important reference value for understanding the evolutionary history of organisms and the kinship relationship between species.
The main characteristics of the mitochondrial genome as a very suitable phylogenetic marker include its small size, its abundance in tissues, the strict direct homology of the genes, the presence of genes or regions with different evolutionary rates, their uniparental inheritance, and the absence of recombination. In addition to the widely used gene sequences, other features of the mitochondrial genome can provide phylogenetic information, including gene content and rearrangement order, changes in the genetic code, or the secondary structure of tRNAs and rRNAs [15]. Internationally, the more well-studied Schmidtea mediterranea and D. japonica are markedly different in their morphological, nuclear, nuclear genomic, mitochondrial genomic, and transcriptomic data [16,17,18,19,20].
To further enrich our limited knowledge of the planarian species, we describe a new species of planarian from a region in southern China, a representative of the genus Dugesia Girard, 1850, on the basis of an integrative approach involving morphological, karyological, molecular data, and mitochondrial genome. The strain has already been successfully preserved and bred in the laboratory. According to the International Code of Zoological Nomenclature and the collection location, we named this new species as Dugesia cantonensis (Guang Yang & Zhinan Yin 2022) [21,22]. This study not only complements new molecular information about the family Dugesiidae in the order Tricladida but also provides a new experimental animal model for comparative research, aiming to enrich the molecular dataset of Dugesiidae species, elucidate their phylogenetic relationships within Tricladida, and establish a viable model organism for evolutionary and developmental studies.
2. Materials and Methods
2.1. Specimen Collection and Culturing
All specimens were collected, using a Pasteur pipette, from submerged leaves in an artificial freshwater pond in Guangzhou, Guangdong Province, China (23°10′48′′ N, 113°22′12′′ E), on 6 April 2022, and were identified based on their external morphology and light brown color pattern. The individuals were collected in a plastic bottle filled with habitat water during transport to the laboratory. The flatworms were maintained in Montjuïc water containing 1.6 mM NaCl, 1.0 mM CaCl_2_, 1.0 mM MgSO_4_, 0.1 mM MgCl_2_, 0.1 mM KCl, and 1.2 mM NaHCO_3_ in a 20 °C constant-temperature incubator in the dark. All animals were fed organic beef liver paste twice per week, and the culture solution was exchanged after each feeding. The flatworms were starved for 1 week before the experiments (being used in karyotype studies, growth curves, histological studies, or DNA extraction). The growth curves of planarians were created by recording the growth of planarians on days 0, 3, 6, and 9 using a digital camera. Thereafter, the body surface area was measured using Image J software version 1.53 and statistically analyzed using GraphPad Prism 9.5.1.
2.2. DNA Extraction, Amplification, Sequencing, and Phylogenetic Analysis
Total genomic DNA was extracted from 10 animals by using the E.Z.N.A. Mollusc DNA Isolation Kit (Omega, Norcross, GA, USA) according to the manufacturer’s protocols. Three gene fragments, 18S ribosomal gene (18S rDNA), 28S ribosomal gene (28S rDNA), and cytochrome C oxidase subunit I (COI), were amplified by polymerase chain reaction (PCR) with the Premix Taq (Takara, Osaka, Japan) according to the reference method [22,23,24]. The following primers were used: 18S rDNA (Forward primer: TACCTGGTTGATCCTGCC AGTAG, Reverse primer: GATCCTTCCGCAGGTTCACCTAC), 28S rDNA (Forward primer: GTCTTGAAACATGGACCAAGG, Reverse primer: GGAACCCCTTCTCCACTTCAGT), and COI (Forward primer: AGCTGCAGTTTTGGTTTTTTGGA, Reverse primer: ATGAGCAACAACATAATAAGTATCATG).
Forward and reverse DNA strands were determined by Sanger sequencing (IGE Biotechnology Co., Ltd., Guangzhou, China). The GenBank accession numbers of the species taxonomic sequences used in the phylogenetic analysis are shown in Supplementary Table S1. Sequence extraction and concatenation was performed using the software PhyloSuite v.1.2.3 [25]. The phylogenetic tree was constructed by using the maximum likelihood method with 1000 boosted replicates under the GTR + I + G model through Mega version 6 [26].
2.3. Mitochondrial Genome and Phylogenetic Analysis
The mitogenome of planarians was sequenced by the next-generation sequencing (NGS) method (Illumina Novaseq 6000, Shanghai, China). The quality of sequencing raw data was evaluated by FastQC and trimmed using Cutatapt software (Galaxy Version 1.16.6) [27]. Obtained Illumina reads were de novo assembled using the NOVOPlasty software version 4.3.1 [28], and the assembled genome was annotated by Mitos2 [29]. This NGS mitochondrial sequencing and linear assembling was completed by Shanghai Jinghan Biotechnology Co., Ltd. (Shanghai, China). We designed primers at both ends of the linear sequence to fill the gaps (F: GCAATTACTTTAGATGTTGCT, R: GCAGCATACATCCATTGCAG). The PCR program was set as follows: denaturation at 94 °C for 1 min, followed by 30 cycles of 98 °C for 10 s, annealing at 55 °C for 15 s, and extension at 68 °C for 5 min. The PCR-amplified products were electrophoresed on a 1% agarose gel, the purified PCR products were ligated with pUC18, and the positive clones were sequenced after transformation (Bio-Transduction Lab Co., Ltd., Shanghai, China). A complete cyclic mitochondrial sequence was finally obtained.
The secondary structures of mitochondrial transfer RNA (tRNA) genes were predicted by tRNAscan-SE (https://lowelab.ucsc.edu/tRNAscan-SE/, accessed on 26 July 2024) and MITOS (http://mitos2.bioinf.uni-leipzig.de/index.py, accessed on 26 July 2024). Circle maps of mitochondria were performed with Organellar Genome DRAW (OGDRAW) version 1.3.1 (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html, accessed on 26 July 2024), The mitochondrial sequences were uploaded to the NCBI website and the mitochondrial circle diagrams were obtained according to the standard method [30].
Mitochondrial phylogenetic analysis was based on complete mitochondrial sequences of free-living flatworms annotated by the NCBI, but the outgroup here was Prosthiostomum siphunculus Delle Chiaje, 1822. Sequence extraction and concatenation were performed using the software PhyloSuite v.1.2.3 [25]. Multiple sequence alignments (MSAs) for protein-coding genes (PCGs) and ribosomal genes were carried out using MAFFT v7.505 [31], and the appropriate nucleotide substitution models were selected by trimming the compared sequences using the software trimAL (version 1.5.0) [32]. The best fit model for each PCG was selected by PartitionFinder2 [33]. Both the maximum likelihood (ML) method and Bayesian inference (BI) method used GTR + I + G models. ML analysis was conducted by IQ-TREE v1.6.2. For BI, MrBayes v3.2.6 was applied with 2,000,000 generations, sampling every 2000 generations, and the tree file was imported into the Interactive Tree of Life (iTOL) website (https://itol.embl.de/, accessed on 8 January 2025) for landscaping. The mitochondrial genome species used for mitochondrial analysis, along with the relevant GenBank accession numbers, are presented in Supplementary Table S2.
2.4. Karyotype Analysis and Nuclear DNA Content
The tail segments of the D. cantonensis (approximately 2.5 mm) were amputated and then incubated for 6h in the dark in a mixture of 0.25% colchicine and 1× Montjuïc water. The colchicine-treated tail segments were then placed in a Petri dish and rinsed with water. The tissue was then punctured with a needle in order to improve permeability, following which the tissue was incubated in deionized water for 20 min at room temperature. The tissue was then placed in fresh Carnot’s fixative (methanol–acetic acid 3:1 dilution) at 4 °C for 30 min. The tail fragment was then placed on a slide and soaked in 60% acetic acid for 5 min. The tissue was then flattened with a coverslip to form a monolayer of cells, after which they were incubated at 4 °C overnight. The slide was then removed and placed on ice, and a blade was used to quickly remove the coverslip. The slide was subsequently brought back to room temperature, and the procedure was repeated three times with 1x phosphate-buffered saline (PBS). Then, the slide was stained with a 1:5000 DAPI solution for 10 min and rinsed with PBS. Then, the slide was sealed and observed under a microscope [34].
Using flow cytometry to estimate the size of the nuclear genome, Zebrafish with known genomes were used as an internal reference species [35], standardized according to the kit provided by the manufacturer (Sysmex, 05-5022, Görlitz, Germany), and then the fluorescence intensity (Sysmex CyFlow^®^ Cube6, Singapore) was tested. The fluorescence intensity can represent the relative content of genomic DNA. The ratio of the DNA content of Zebrafish and flatworms can be derived from the fluorescence peaks of the tested sample and the PI-DNA complex of the Zebrafish sample, which can be multiplied by the C-value of the Zebrafish to calculate the C-value of the tested D. cantonensis [36]. D. cantonensis DNA content = [(D. cantonensis sample peak mean)/(Zebrafish standard peak mean)] × Zebrafish standard DNA content.
2.5. Histology
Following 7d of starvation, worms were euthanized using 5% HCl prior to fixation. After fixation in Bouin’s Fixative Solution (saturated nitric acid–formaldehyde–glacial acetic acid = 68:25:7), the worms were rinsed with 70% ethanol and then dehydrated in an ascending series of ethanol baths, cleared in terpineol, and embedded in wax (Paraplast Plus, Sigma, Saint Louis, MO, USA). Serial sections were prepared at 6 μm intervals and stained with hematoxylin and eosin. This was followed by visualization under a microscope (Leica, Wetzlar, Germany) [37]. Histological preparations of specimens have been deposited in the Laboratory of Department of Pathogen Biology, School of Medicine, Jinan University.
3. Results
3.1. Molecular Phylogeny of 18S rDNA, 28S rDNA, and COI
In order to identify the species of D. cantonensis, we used the maximum likelihood method to build a phylogenetic tree based on the concatenated nucleotide sequences of 18S rDNA (GenBank accession no. PQ901321), 28S rDNA (GenBank accession no. PQ901322), and COI (GenBank accession no. PV076738), and we ascertained the phylogenetic relationships among a total of 38 species within the family Dugesiidae. The phylogenetic tree showed a 30–100% level of support at all nodes. Within the broader taxonomic classification of the Dugesiidae family, S. mediterranea, Schmidtea polychroa, and Recurva postrema were chosen as the outgroup taxa [38]. The phylogenetic tree showed that D. cantonensis, Dugesia sp.a ZY 2023 (D. ancroaria), and D. notogaea form a clade. In this clade, the branch between D. cantonensis and D. ancroaria had support values (bootstrap) of 99%, and the common closest branch of D. notogaea had a value of 100% (Figure 1).
3.2. Mitochondrial Genomic Features and Phylogenetic Analyses
In order to further ascertain the taxonomy of D. cantonensis, complete mitochondrial genome sequencing is carried out (GenBank accession no. PV083436). Its mitochondrial genome is 18,125 bp, which is a relatively larger size when compared with that of S. mediterranea at 17,176 bp (GenBank accession no. KM821047) and D. japonica at 17,799 bp (GenBank accession no. AB618487). The mitochondrial genome is composed of 36 genes (all genes are encoded in the same direction) consisting of 12 protein-coding genes, 22 tRNA genes, and 2 ribosomal RNA genes but lacking the subunit 8 of ATP synthase (atp8) gene, which is a common feature in flatworm mtDNA [39,40,41] (Figure 2A). The mitochondrial genome of D. cantonensis also exhibits deviations in base composition and skew (unequal representation of complementary bases on the same strand). Its composition is 23.3% A, 8.2% C, 15.8% G, and 52.6% T, and it is enriched in AT (A + T content of 75.9%), with T being the most common base on the coding strand. For the entire sequences, AT skew and GC skew are −0.31 and 0.32, respectively, when calculated using the formulae [42]. The lower value of AT skewness in these genes is generally considered to be associated with the formation of stem–loop secondary structures (i.e., base pairing between A and T in the stem regions of their corresponding secondary structures) [18,39].
The phylogenetic tree is constructed from concatenated protein-coding genes of the mitochondrial genome. The tree is composed of three families, Prosthiostomidae, Geoplanidae, and Dugesiidae of Platyhelminthes, which possess the annotation of a complete mitochondrial genome in NCBI. It uses P. siphunculus as an outgroup taxon. According to the phylogenetic tree, D. cantonensis belongs to the clade of the family Dugesiidae. The phylogenetic tree reveals that D. cantonensis, D. ryukyuensis, Girardia tigrina, and Girardia sp. constitute a clade, and this clade is comparatively close to D. japonica and S. mediterranea. The mitochondrial genome of D. cantonensis is more closely related to the Girardia genus, yet morphologically, its auricles are very different from those of the Girardia genus, which has pointed auricles (Figure 2B). The classification based on morphology and mitochondrial sequences clashes. We have given priority to the identification of specimens with obvious morphological differences [43].
3.3. Taxonomy
Systematicaccount
Order Tricladida Lang, 1884 *
Suborder Continenticola Carranza, Littlewood, Clough, Ruiz-Trillo, Baguñà & Riutort, 1998 *
Family Dugesiidae Ball, 1974 *
Genus Dugesia Girard, 1850 * D. cantonensis Guang Yang & Zhinan Yin, sp.nov.2022****Zoobank registration: urn: lsid: zoobank.org: pub: D04C1C71-0B3B-4EB5-95F3-9A40635090ED.Material examined: an artificial freshwater pond, Guangzhou city, Guangdong Province, China, 23°10′48′′ N, 113°22′12′′ E, 6 April 2022, coll.Guang Yang and co-workers, sagittal section on 15 slides.Etymology: The specific epithet cantonensis (Latin adjective, nominative singular masculine) originates from the historical exonym Canton (Guangzhou, China), combined with the Latin suffix -ensis (pertaining to). This nomenclature reflects the geographic origin of this species, which was first discovered in freshwater ecosystems of the Guangzhou region.Diagnosis: D. cantonensis is characterized by a triangular head with a blunt, rounded tip accompanied by short, flattened auricles. The dorsal surface of the body has a light brown pigmentation with the body separated by a weakly pigmented dorsal midline stripe. The pharynx is located in the anterior of the body and is marked by a distinct white area. As a diploid organism, it possesses 16 chromosomes and a nuclear genome size of 2.5 Gb. The mitochondrial gene arrangement is as follows: cox1-E-nad6-nad5-S2-D-R-cox3-I-Q-K-atp6-V-nad1-W-cox2-P-nad3-A-nad2-M-H-F-rrnS-L1-Y-G-S1-rrnL-L2-T-C-N-cob-nad4l-nad4.
3.4. Description
D. cantonensis is elongated and constricted with a bluntly rounded anterior end. The posterior segment is a thin, conical tail end. Adult worms are about 1.0–1.2 cm long and light brown in color. The length of the pharynx is roughly one-tenth of the body length: the root position of the oropharyngeal is at around one-quarter of the anterior, while the oral opening is at approximately one-third of the anterior. The head and auricle are less pigmented. The two small eyespots are located near the anterior end and are oval in shape. The collection environment of D. cantonensis is shown in Figure 3A. There are no sexual organs in the posterior region (Figure 3B,C). In order to examine the regenerative capabilities of various body parts, D. cantonensis had their (trunk and tail) and head amputated (Figure 3D); planarians had their (head, trunk) and tail amputated (Figure 3E); and planarians had their trunk and (head and tail) amputated (Figure 3F). The whole regeneration process of the head, trunk, and tail resulted in complete regeneration by the 9th day. From a single D. cantonensis, it was propagated to approximately 100,000 individuals via amputation within 3 years, and tripling the time it stayed stable, suggesting the identification and nurturing of a new monoclonal planarian strain, D. cantonensis.
3.5. Karyology and Nuclear Genome Size Assay
In addition to molecular information, karyotype analysis and nuclear genome size are important complementary methods for species identification. Chromosomal measures and the population of individuals from the planarians were compiled [10]. As can be observed, the morphological structure of the chromosomes is obvious, with clearer mitoses, which facilitate karyotyping. D. cantonensis was diploid, with a chromosome complement of 2n = 16. Following the chromosome division criteria for determining the location of the chromosomes [44], chromosome 5 is found to be of the submetacentric type and the rest of the chromosomes are of the metacentric type. Karyotype parameters, including the relative length, arm ratio, and centromeric index, are given in Supplementary Table S3. A chromosomal plate and idiogram of the karyotype are shown in Figure 4A,C.
For nuclear genome size assay, the special fluorescent dye PI is used to bind to the DNA in the worm cells, and the cells dyed with the fluorescent dye emit fluorescence under laser irradiation. Utilizing the Zebrafish genome as an internal reference, which has an approximately 1.5 giga bp (Gb) haploid nuclear genome [45], the 2957 events were counted, with a gated cell rate of about 75.74% and a geometric mean of 26,167.11 in Zebrafish. D. cantonensis counted 3124 events with a gated cell rate of about 69.98% and a geometric mean value of 44,463,863. The results demonstrated that the size of the planarian genome is approximately 2.5 Gb. The size of the nuclear genome of D. japonica is 1.13 G and that of S. mediterranea is 773.9 Mb [17,19]. D. cantonensis has a relatively larger genome content with developmental complexity in its evolutionary position. In the phylum Platyhelminthes, the reduction in the nuclear genome is achieved by compression and gene loss as the organism moves from free-living to parasitic life [46], with the replication of smaller genomes or the transcription/translation of fewer genes requiring less energy, thus freeing up resources for other functions [47]. Variation in genome size can constrain, and simultaneously be constrained by, the evolution of the organism in which it finds itself [48] (Figure 4D,E).
3.6. Histology
To observe the organs of the planarian, histological examinations are carried out. After the worms have been stained, the structure of the organs is clearly visible (Figure 5A). In the head of the sagittal section, the structure of the eye point and the brain area can be clearly seen (Figure 5B). The structure of the planarian pharynx is clearly delineated, and above the pharynx is the intestinal duct. The entire pharynx is within the pharyngeal cavity and appears contracted. The pharyngeal lumen is located inside the pharynx (Figure 5C). The muscles and digestive tract are visible, but no reproductive structures such as seminal vesicle, copulatory bursa, vitelline gland, ejaculatory ducts, and oviducts are found (Figure 5D–F).
3.7. Protein-Coding Gene Preference Analysis and Transfer Ribosomal RNA of the Mitogenome
The Relative Synonymous Codon Usage (RSCU) value can measure the frequency of codon usage. As shown in Figure S1, the three codons with the higher RSCU values in the mitochondrial genomes of the D. cantonensis were UCU, GCU, and ACU, indicating that these three codons were the most frequently used in encoding the mitochondrial genome (Figure S1). The numbers on the bar graph represent the frequency of codon usage.
All 22 tRNAs were detected, with 21 exhibiting the typical cloverleaf structure. However, the circular structure of the glutamate tRNA was atypical, and the four stems characteristic of the cloverleaf structure were missing. This suggests that gene mutations may have occurred during the long-term evolutionary process. The secondary structures of the 22 tRNAs from the D. cantonensis newly sequenced mitogenomes were generated and are presented in Figure S2.
4. Discussion
The identification and nurturing of D. cantonensis represent a significant contribution to freshwater planarian biodiversity and evolutionary studies. The species exhibits unique morphological traits (e.g., pharyngeal position at the anterior quarter of the body), a larger mitochondrial genome (18,125 bp), a nuclear genome size of 2.5 Gb, and a diploid karyotype (2n = 16). These characteristics not only expand the taxonomic diversity of the genus Dugesia, but also provide a potential model for exploring genome evolution in free-living flatworms. The establishment of a stable culture stain facilitates research on regeneration, environmental adaptation, and developmental mechanisms.
The genus Dugesia exhibits a high degree of geographic population diversity on a global scale. This diversity is attributable to genetic differentiation resulting from geographic isolation, which in turn leads to the formation of new species of planarians [49,50]. However, the classification of freshwater planarians is challenging, and the limited availability of taxonomic keys to identify species, or the fact that most descriptions are old and based only on external morphological features, complicates the classification of some freshwater planarians [51]. The application of internal anatomical characteristics has resulted in alterations to the classification system over time [6]. In most instances, the absence of synapomorphies that define species or higher level taxonomic groups precludes the use of morphological features for the purpose of inferring phylogenetic relationships. Consequently, their classification is not based on their natural groupings as deduced from a phylogeny [52,53,54]. The use of molecular data to infer phylogenies has been pivotal for understanding the origin and evolution of numerous platyhelminth features, and molecular markers have become a vital tool for elucidating a plethora of diversity in many cases not, or only partially, predicted by morphological appearance [52]. This study utilized an integrative approach combining morphological, molecular, and cytogenetic data to validate D. cantonensis as a new species.
Nuclear genome sequencing remains critical but has not been fully explored, mainly because of its high cost and difficulty in assembly, especially in planarians, where few reference genomes are available and current taxonomic standards do not generally require nuclear genome data. However, nuclear genome data may increasingly complement traditional methods of species identification. The initial analysis at the higher level of taxonomy was conducted using 18S rRNA, with the subsequent addition of the mitochondrial gene COI, which has proven invaluable for intraspecific comparisons and for distinguishing closely related species [55,56]. In this study, we carried out phylogenetic analysis by means of complete mitochondrial sequences as well as 18S, 28S, and COI concatenated genes, and the results of the analyses were quite different from the evolutionary distances of S. mediterranea and D. japonica. The 18S, 28S, and COI exhibited high levels of saturation, which obscured the weak signals of other genes in the basal branches when working at the family level, as evidenced by the Continenticola phylogeny [24].
The complete mitochondrial genome offers a multitude of molecular markers that are conducive to the investigation of a variety of biological characteristics, including the impact of divergent ecological adaptations [57]. Moreover, the phylogenetic relationships among populations or species can be elucidated. This is because mitochondrial DNA usually does not recombine, frequently shows neutral evolution, and mt markers have smaller effective population sizes than their nuclear counterparts, which result in shorter coalescent times [58,59]. These features make mtDNA especially appropriate for either phylogeographical or population genetic studies [60]. In this study, we found that the size of the mitochondrial genome of D. cantonensis is relatively large when compared with that of S. mediterranea, D. japonica, and related *Dugesia ancoraria * [3].
Some freshwater planarians (Platyhelminthes, Turbellaria, Seriata, and Tricladida) switch between asexual and sexual reproduction [61,62]. The asexual individuals reproduce by fission without forming any sexual organs, whereas the sexual ones are hermaphroditic [63]. We sliced D. cantonensis by HE staining techniques. D. cantonensis have no obvious reproductive system in histology and are asexual worms. In a large number of observations regarding the behavioral science of planarians, the asexual species reproduces solely by fission and exhibits no sexuality whatsoever. Recent studies have shown that D-amino acids can be utilized to transform asexual worms into sexual worms [64]. Diploid organisms in D. cantonensis, which possess paired chromosomes, hold great potential in the shift towards sexuality. Gender transition is a scientific issue worthy of further study.
In addition, along with the progress of omics technologies, clarifying the biology of D. cantonensis from the perspectives of whole-genome sequencing, epigenomics, single-cell omics, metabolomics, and spatial transcriptomics will be advantageous and obtain more in-depth understandings of the development and pathogenesis of parasitic helminths.
Overall, we identified a new species of planarian and verified our findings by employing a range of techniques. The outcomes not only affirm the uniqueness of D. cantonensis but also provide insights into its evolutionary associations within the genus Dugesia. This discovery broadens the recorded diversity of planarians and presents a new strain for comparative studies.
5. Conclusions
In this study, we identify and describe a new asexual freshwater planarian species from southern China, D. cantonensis, using an integrative approach that combines morphological, karyotypic, molecular phylogenetic, and mitochondrial genomic analyses. The species displays distinctive morphological and genetic characteristics, exceptional regenerative capacity, and stable long-term asexual reproduction. Its mitochondrial and nuclear genomes suggest unique evolutionary and ecological adaptations. This discovery expands the known diversity of Dugesiidae in East Asia and offers a valuable model for research on regeneration, genome evolution, and asexuality.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Little C. The Colonisation of Land: Origins and Adaptations of Terrestrial Animals Cambridge University Press Cambridge, UK 1983
- 2Chen G.-w. Wang L. Wu F. Sun X.-j. Dong Z.-m. Sluys R. Yu F. Yu-Wen Y.-Q. Liu D.-z. Two new species of Dugesia (Platyhelminthes, Tricladida, Dugesiidae) from the subtropical monsoon region in Southern China, with a discussion on reproductive modalities BMC Zool.202272510.1186/s 40850-022-00127-837170346 PMC 10126995 · doi ↗ · pubmed ↗
- 3Zhu Y. Huang J. Sluys R. Liu Y. Sun T. Wang A.-T. Zhang Y. Integrative description of a new species of Dugesia (Platyhelminthes, Tricladida, Dugesiidae) from southern China, with its complete mitogenome and a biogeographic evaluation Zoosystematics Evol.202410016718210.3897/zse.100.114016 · doi ↗
- 4Wang L. Wang Y. Dong Z. Chen G. Sluys R. Liu D. Integrative taxonomy unveils a new species of Dugesia (Platyhelminthes, Tricladida, Dugesiidae) from the southern portion of the Taihang Mountains in northern China, with the description of its complete mitogenome and an exploratory analysis of mitochondrial gene order as a taxonomic character Integr. Zool.202217119312143478315310.1111/1749-4877.12605 · doi ↗ · pubmed ↗
- 5Song X.-Y. Li W.-X. Sluys R. Huang S.-X. Li S.-F. Wang A.-T. A new species of Dugesia (Platyhelminthes, Tricladida, Dugesiidae) from China, with an account on the histochemical structure of its major nervous system Zoosystematics Evol.20209643144710.3897/zse.96.52484 · doi ↗
- 6Kawakatsu M. Oki I. Tamura S. Sugino H. Studies on the morphology, karyology and taxonomy of the Japanese freshwater planarian Dugesia japonica Ichikawa et Kawakatsu, with a description of a new subspecies, Dugesia japonica ryukyuensis subspec. nov Bull. Fuji Women’s Coll.19761481126
- 7Sluys R. Kawakatsu M. Contribution to an inventory of the freshwater planarians of Australia and New Zealand (Platyhelminthes, Tricladida, Dugesiidae), with distribution maps of the species examined Beaufortia 200151163198
- 8Hind K.R. Saunders G.W. Molecular markers from three organellar genomes unravel complex taxonomic relationships within the coralline algal genus Chiharaea (Corallinales, Rhodophyta)Mol. Phylogenetics Evol.20136752954010.1016/j.ympev.2013.02.02223467004 · doi ↗ · pubmed ↗