Colorectal Cancer Biomarker Identification via Joint DNA-Methylation and Transcriptomics Analysis Workflow
Olajumoke B. Oladapo, Marmar R. Moussa

TL;DR
This paper identifies potential biomarkers for colorectal cancer by combining DNA methylation and gene expression data from multiple datasets.
Contribution
A novel computational workflow integrating DNA methylation and transcriptomics data to identify methylation-regulated biomarkers in colorectal cancer.
Findings
150 methylation-regulated genes (MRGs) were identified, with GNG7 and PDX1 common across all cohorts.
Functional analysis highlighted key pathways like Wnt signaling and extracellular matrix organization in CRC.
The study demonstrates the effectiveness of an in silico approach for biomarker discovery in colorectal cancer.
Abstract
Background: Colorectal cancer (CRC) is a term that refers to the combination of colon and rectal cancer as they are being treated as a single tumor. In CRC, 72% of tumors are colon cancer, while the other 28% represent rectal cancer. CRC is a multifactorial disease caused by both genetic and epigenetic changes in the colon mucosal cells, affecting the oncogenes, DNA repair genes, and tumor suppressor genes. Currently, two DNA methylation-based biomarkers for CRC have received FDA approval: SEPT9, used in blood-based screening tests, and a combination of NDRG4 and BMP3 for stool-based tests. Although DNA methylation biomarkers have been explored in colorectal cancer (CRC), the identification of robust and clinically valuable biomarkers remains a challenge, particularly for early-stage detection and precancerous lesions. Patients often receive diagnoses at the locally advanced stage,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
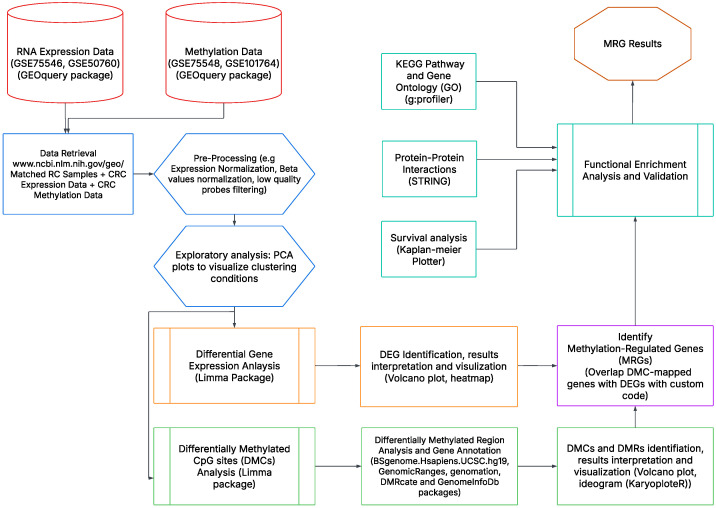 Figure 1
Figure 1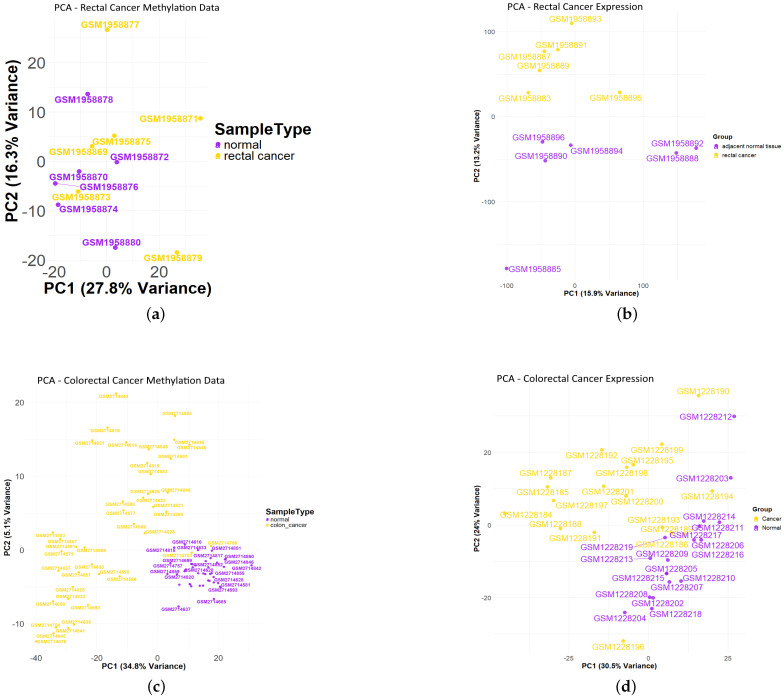 Figure 2
Figure 2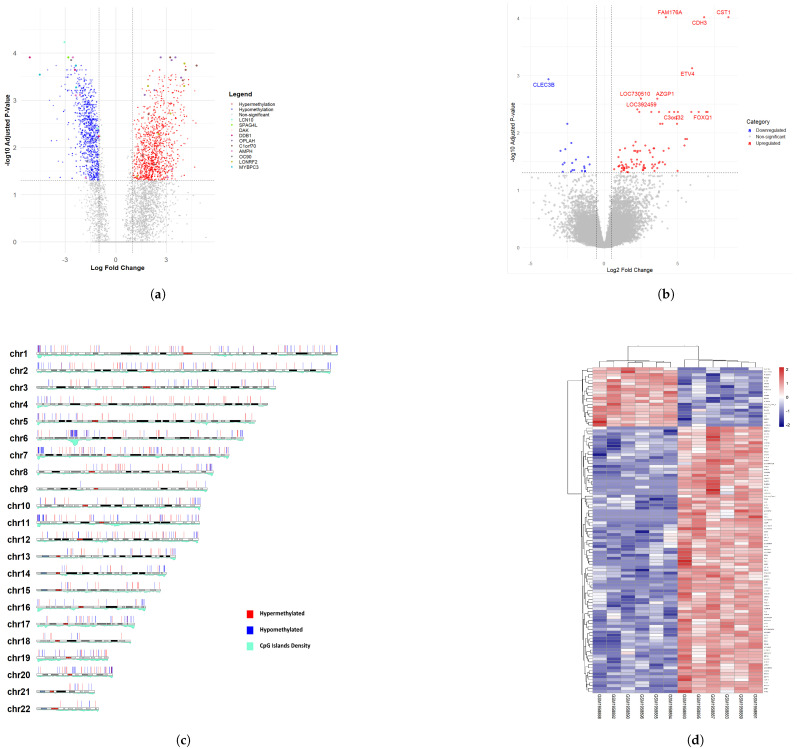 Figure 3
Figure 3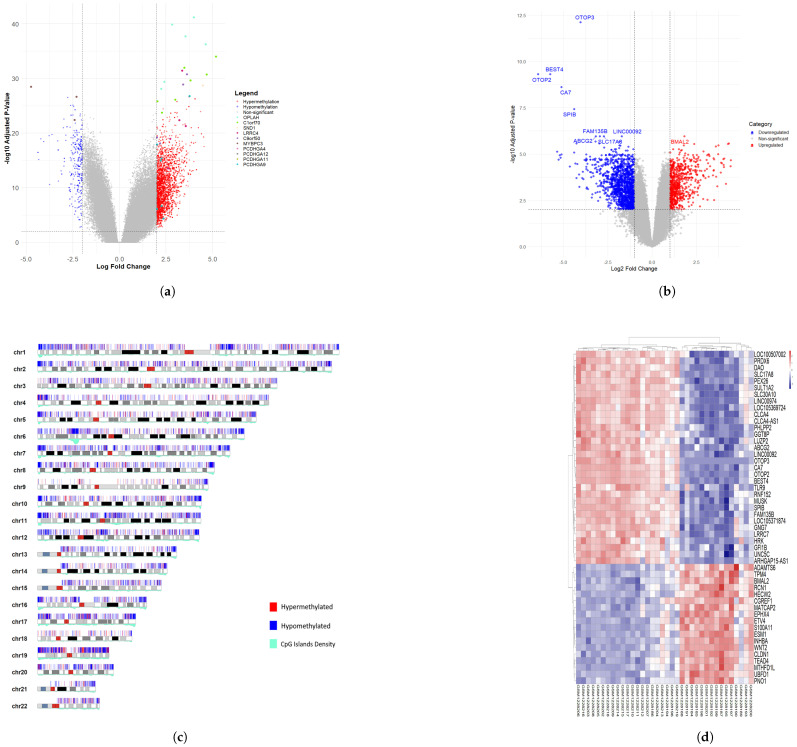 Figure 4
Figure 4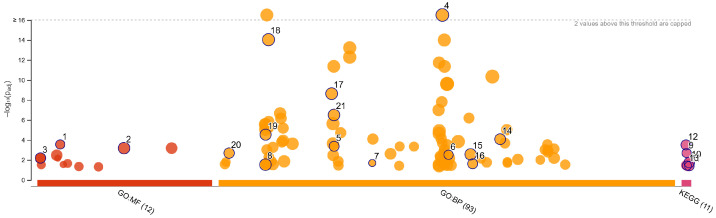 Figure 5
Figure 5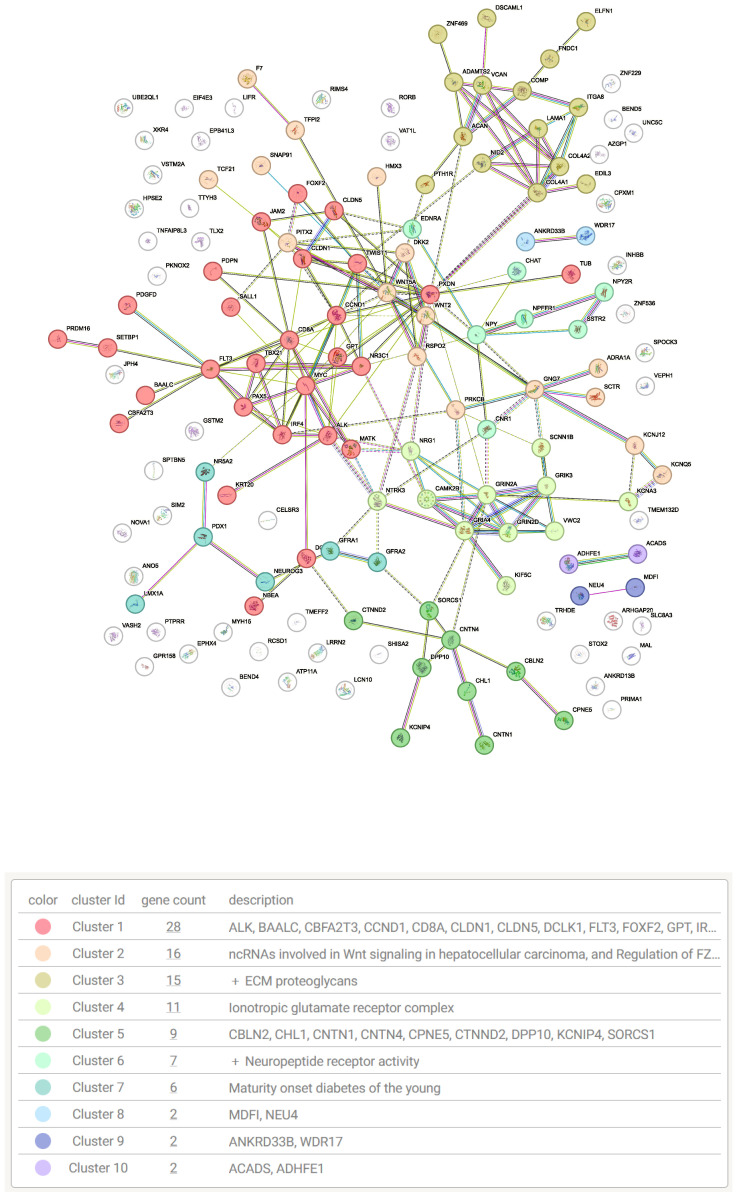 Figure 6
Figure 6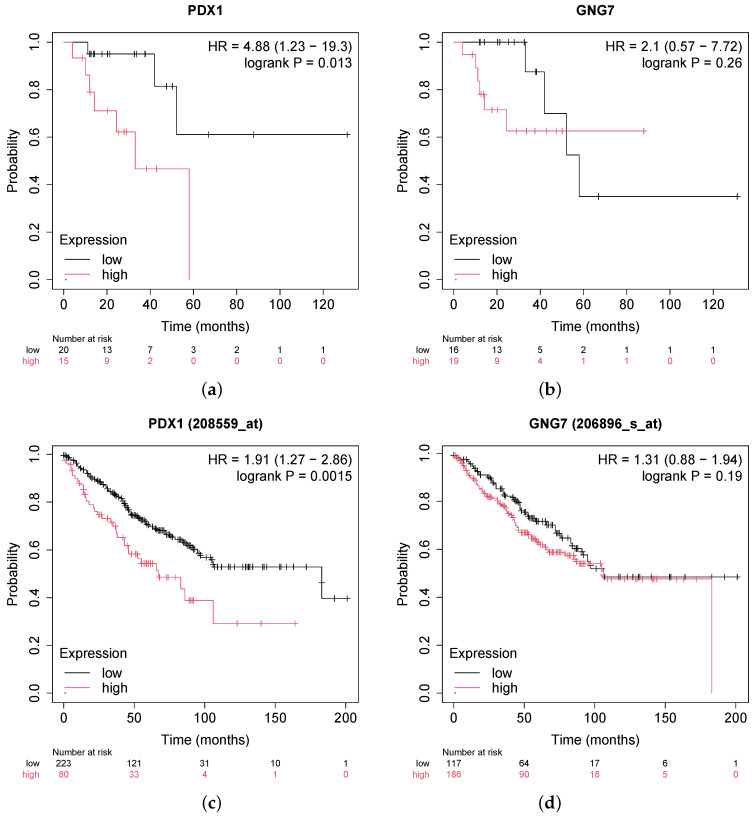 Figure 7
Figure 7- —NSF
- —NIH
- —OU-BIC2.0
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFerroptosis and cancer prognosis · Cancer-related molecular mechanisms research · Epigenetics and DNA Methylation
1. Introduction
Colorectal cancer (CRC) is a term that refers to the combination of colon and rectal cancers, which are treated as a single tumor. In CRC, 72% of tumors are colon cancer, while the remaining 28% are rectal cancer [1]. Colorectal cancer ranks as the third most prevalent cancer and the second largest cause of mortality, with an anticipated incidence rate exceeding 60% by 2030 [1,2]. The 5-year survival rate for 90% of patients diagnosed with CRC in early and localized stages is significantly higher than the 13.1% rate observed in advanced stages and metastatic cases [3]. Early detection is essential for the survival of patients diagnosed with CRC, and biomarkers are pivotal in its diagnosis and prognosis. However, only a limited number of biomarkers have been integrated into clinical practice, underscoring the necessity to develop additional biomarkers in CRC [4]. Currently, microRNAs, DNA mutations, methylation, proteins encompassing various epigenetic functions, and gut microbiomes are areas investigated for the identification of CRC biomarkers [5].
DNA methylation patterns in normal and tumor-specific cells exhibit markedly distinct profiles, which can facilitate the identification of DNA from tumor samples, hence serving as a promising biomarker [6]. Currently, two DNA methylation biomarkers for colorectal cancer (CRC) have been approved by the FDA: SEPT9, utilized in blood screening tests, and a combination of NDRG4 and BMP3 for stool tests [7]. Recently, many studies have introduced promising DNA methylation biomarkers for CRC. Shen et al. [7] identified two potential CpG site biomarkers for colorectal cancer: cg13096260 and cg12993163, from 76 pairs of CRC and adjacent normal tissue samples, 348 stool samples, and 136 blood samples. In a similar manner, the Stool ColoDefense test used by Zhao et al. [8] found the DNA methylation of SEPT9 and SDC2 as a composite biomarker for CRC. Despite the investigation of DNA methylation biomarkers in CRC, the discovery of reliable and clinically significant biomarkers continues to pose a problem, especially for early-stage detection and precancerous lesions. Current biomarkers frequently exhibit insufficient sensitivity and specificity for early detection, resulting in patients typically being diagnosed at a locally advanced stage, which constrains their potential application in clinical environments [4,9].
This study seeks to fill this gap by utilizing a bioinformatics pipeline to find novel DNA methylation-regulated genes linked to CRC. This study employs the methodology established by Li et al. [10], who identified methylation-regulated genes in varicose vein disease and classified these genes as biomarkers for varicose vein disease alongside their traits which were taken into consideration in their analysis. In this study, we aim to identify methylation-regulated genes in CRC samples by analyzing publicly accessible methylation and expression datasets of CRC for candidate biomarkers that demonstrate consistent epigenetic modifications in CRC samples. The primary objective is to identify biomarkers that may be subsequently validated for their diagnostic and prognostic capabilities in CRC and precancerous lesions.
2. Materials and Methods
In the following section, we describe the main components of our joint analysis workflow. An overview of the workflow processes is shown in Figure 1.
2.1. Data Collection
The datasets utilized in this study were retrieved from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/, accessed on 13 May 2025) using the GEOquery package [11]. Specifically, datasets GSE75548 and GSE75546 were obtained for rectal cancer, consisting of matched patient samples. GSE75548 represents expression profiling by microarray, whereas GSE75546 represents methylation profiling by genome tiling array; both datasets contain six paired samples of rectal cancer and corresponding normal tissues. Additionally, datasets GSE50760 (expression profiling by high-throughput sequencing) and GSE101764 (methylation profiling by microarray) were employed for colon cancer analyses. The GSE50760 dataset was subsetted to retain only colon cancer samples, yielding a total of 36 samples. The GSE101764 dataset was filtered to include paired samples from patients aged 40 and above, resulting in a total of 82 samples, thus ensuring consistency in biological characteristics, including age, across all analyses. In total, 130 samples are analyzed in this study; Figure 2 summarizes the samples in both expression and methylation data via principal component analysis.
2.2. Identifying and Mapping Differentially Methylated CpG Sites
Differentially methylated CpG sites (DMCs) between normal and cancer tissue samples were identified using the Limma package [12]. For rectal cancer, DMCs with Adj.P.Value < 0.05 and were considered statistically significant. Due to the larger sample size available for colon cancer analyses, more stringent thresholds were applied, with significance defined by an Adj.P.Value < 0.01 and . Differentially methylated regions (DMRs) between normal and cancer samples were identified using the DMRcate package [13], with a false discovery rate (FDR) threshold of 0.001. DMRs were defined as genomic regions containing at least two significant DMCs (C = 2) within a 1000 bp window ( ). Genomic coordinates of identified regions were validated using the BSgenome.Hsapiens.UCSC.hg19 package [14], ensuring the inclusion of only standard autosomal chromosomes. Subsequently, DMCs obtained from Limma were cross-referenced with the DMR results, classifying them into hypermethylated, hypomethylated, or non-significant categories. A karyogram visualizing hypermethylated (red) and hypomethylated (blue) genomic regions was generated using the karyoploteR package [15].
2.3. Normalization and Filtering
Expression data for rectal cancer were obtained in a pre-normalized form from the GEO archive and further filtered using median expression values. For colon cancer, the normalization and filtering of samples were performed using EdgeR [16], ensuring minimal batch effect in expression and methylation data (see Supplementary Figure S1).
2.4. Identification of Differentially Expressed Genes (DEGs)
Differentially expressed genes (DEGs) between normal and cancer tissue samples were identified using the Limma package [12]. For rectal cancer, DEGs with Adj.P.Value < 0.05 and were considered statistically significant. Due to the larger sample size available for colon cancer analyses, more stringent thresholds were applied, with significance defined by an Adj.P.Value < 0.01 and . The results were visualized using a volcano plot to highlight upregulated, downregulated, and non-significant genes.
2.5. Statistical Methods for Differential Analyzes
As discussed in previous sections, Limma R package and algorithms were used to calculate differential expression (or methylation). To summarize, this method apply fitting linear models to normalized expression data, considering factors like inter-gene correlation and precision weights. The method then compares the expression levels of different groups or conditions using t-tests, identifying genes with significant differences.
2.6. Identification and Analysis of Methylation-Regulated Genes (MRGs)
Gene symbols from the annotated DMRs were compared with significantly differentially expressed genes (DEGs). This integration identified common genes: methylation-regulated genes (MRGs) that showed both methylation alterations and differential expression patterns.
2.7. Validation and Functional Enrichment of Methylation-Regulated Genes
Biological processes and pathways associated with methylation-regulated genes (MRGs) were identified using Gene Ontology (GO) and KEGG pathway enrichment analysis performed using g:Profiler [17] methods. Protein–protein interaction (PPI) networks were constructed using the STRING database [18] to identify gene clusters and their associated functional and regulatory pathways, further validating the methylation-based regulation of genes. Additionally, survival analysis (Kaplan–Meier (KM) [19] overall survival (OS) method) was conducted on select genes using clinical data from colon and rectal cancer patients to evaluate the prognostic significance of two spotlight MRGs.
3. Results
3.1. Differentially Methylated CpG Sites (DMCs) and Differentially Expressed Genes (DEGs) of Rectal Cancer Cohort
The methylation and expression datasets of rectal cancer were analyzed to identify differentially methylated CpG sites (DMCs) by fitting a generalized linear model from limma. The results from the DMC analysis were cross-referenced with differentially methylated regions (DMRs) to enhance the reliability of the findings. A total of 678 genes were classified as significantly hypermethylated or hypomethylated within the identified DMRs. Differential expression analysis revealed 101 genes that were significantly up- or downregulated in rectal cancer. The lists of significant genes from the methylation and expression analyses are provided in Supplementary Tables S1 and S2, respectively. Figure 3 illustrates the volcano plots for DMCs and DEGs, the karyogram highlighting DMRs with significant hypermethylation and hypomethylation, and a heatmap of the top 50 differentially expressed genes.
3.2. Differentially Methylated CpG Sites (DMCs) and Differentially Expressed Genes (DEGs) of Colon Cancer Cohort
Conserving the methodology applied to the rectal cancer cohort, we extended this analysis to the colon cancer datasets. The methylation and expression data of the colon cancer samples were analyzed to identify differentially methylated CpG sites (DMCs) by fitting a linear model from limma. The results from the DMC analysis were cross-referenced with differentially methylated regions (DMRs) to enhance the reliability of the findings. A total of 1053 genes were classified as significantly hypermethylated or hypomethylated within the identified DMRs. Differential expression analysis revealed 2130 genes that were significantly upregulated or downregulated in the colon cancer group. The lists of significant genes from the methylation and expression analyses are provided in Supplementary Tables S3 and S4, respectively. Figure 4 illustrates the volcano plots for DMCs and DEGs, the karyogram highlighting DMRs with significant hypermethylation and hypomethylation, and a heatmap of the top 50 differentially expressed genes.
3.3. Methylation-Regulated Genes (MRGs)
Out of the 678 unique genes identified from both the DMC and DMR analyses in rectal cancer, six genes that overlapped with the 101 differentially expressed genes (DEGs). Similarly, 146 overlapping genes were identified from the colon cancer DMC and DMR analyses with corresponding DEGs. In total, 150 genes were inferred as methylation-regulated genes (MRGs) across the total colorectal cancer cohort, with two genes in particular—PDX1 and GNG7—consistently identified in both rectal and colon cancer individual analyses. These common genes were considered as promising candidates of MRGs. To validate the identified MRGs, we conducted further functional annotation, pathway enrichment, and survival analysis, highlighting the role of these genes in CRC. A select group of the identified MRGs, along with their fold change, average expression, and adjusted p-values, is summarized in Table 1, which highlight all genes pertaining to rectal cancer cohort (six genes in total) and the top 10 MRGs from colon cancer; the full list includes the shared genes (highlighted in bold) across all samples.
3.4. Validation and Functional Enrichment of Methylation-Regulated Genes
The methylation-regulated genes (MRGs) were further subjected to functional enrichment analysis using KEGG and Gene Ontology databases via g:Profiler methods [19]. Key biological pathways identified from the enrichment results include the Wnt signaling pathway, pathways in cancer, and extracellular matrix organization, among others. Additionally, several neurogenesis and neuron development pathways were identified, highlighting the role of the nervous system (enteric nervous system) in the etiology and development in CRC. Figure 5 presents the enrichment and functional analysis results, highlighting the top enriched pathways associated with MRGs. Table 2 summarizes the functional pathways associated with the methylation-regulated genes (MRGs), as identified through Gene Ontology and KEGG pathway enrichment analysis.
In addition, Figure 6 illustrates the protein–protein interaction (PPI) network generated using the STRING database, along with functionally relevant pathways derived from these interactions.
Furthermore, we performed additional validation through survival analysis performed on public clinical data for two selected highlighted genes, PDX1 and GNG7—which were commonly identified in both datasets. This analysis is presented in Figure 7. These Kaplan–Meier plots illustrate the association between gene expression levels and patient survival across rectal and colon cancer cohorts.
4. Discussion
Colorectal cancer (CRC) arises when the normal epithelial cells of the colon and rectum undergo transformation into a precancerous lesion, ultimately progressing to an advanced carcinoma capable of metastasizing to other organs [1]. The risks of developing colorectal cancer (CRC) are associated with age, environmental influences, behavioral patterns, and genetic determinants [20]. Raut et al. [21] identified two fecal DNA methylation biomarkers for detecting stages in colorectal cancer (CRC). Bach et al. [22] discovered SEPT9 and SDC2 as critical markers for non-invasive colorectal cancer (CRC) detection by urine-based DNA methylation analysis. DNA methylation has been extensively studied in CRC; Huang et al. [23] identified distinct tumor clusters with methylated CpG islands linked to metabolic pathways, enhanced ATP production, and tumor aggressiveness in CRC.
In this current study, we analyzed data from a publicly available dataset on colon and rectal cancer samples and carried out differential methylation and expression analysis on these datasets. We identified significant hypermethylated and hypomethylated genes in CRC and found genes that were methylation-regulated suggesting methylation plays a role in the alterations of these gene expression patterns. Similarly, Miao et al. [24], through an integrated analysis in the pathogenesis of coronary artery disease, found overlaps between differentially methylated genes (DMGs) and DEGs through their intersection and carried out subsequent analysis to highlight genes important in the pathogenesis of coronary heart disease. Sun et al. [25], through an integrated analysis, identified eight genes that are regulated by methylation and proposed these genes to have therapeutic and diagnostic relevance in lung cancer.
A total of 150 genes were identified as MRGs from CRC analysis which includes PDX1 and GNG7 as spotlight genes were consistently found in rectal as well as colorectal cancer samples in both differentially expressed and methylated gene groups.
Findings from Liu et al. [26] showed 411 upregulated genes that were significantly hypomethylated and 239 downregulated genes that were hypermethylated. The hub genes that can serve as important biomarkers for CRC. Similarly, Sun et al. [27] identified hub genes that were differentially expressed in CRC analysis and suggested these hub genes as biomarkers of CRC.
In this study, we identified 101 and 2130 significant differentially expressed genes (DEGs) in rectal and colon cancer, respectively. Correspondingly, 678 and 1053 significant differentially methylated CpG sites (DMCs) were detected in rectal and colon cancer. By intersecting the DEGs and DMCs from each dataset, we identified a total of 150 methylation-regulated genes (MRGs). Notably, PDX1 and GNG7 were common to both rectal and colon cancer analyses, with GNG7 also ranking among the top ten genes in colon cancer.
GNG7, a component of heterotrimeric G proteins, is highly enriched in the striatum and plays a crucial role in the neuroprotective response mediated by A2A adenosine and D1 dopamine receptors. Previous studies have reported GNG7 downregulation in various cancers, including pancreatic, gastrointestinal tract, renal, and lung cancers [28]. In our study, we identified GNG7 as being downregulated and hypomethylated in colorectal cancer. PDX1 is predominantly expressed in the islets of Langerhans, central nervous system, and gastrointestinal tract [29,30]. It is a critical transcription factor involved in pancreas development and has been implicated in colorectal cancer (CRC). A recent study by Lee et al. [31] reported that the hypermethylation of PDX1 serves as a potential biomarker for CRC prognosis [31]. Consistent with these findings, our current analysis also demonstrates that PDX1 is hypermethylated and correspondingly upregulated in colorectal cancer samples.
We performed KEGG pathway enrichment and Gene Ontology (GO) analyses using g:Profiler, alongside protein–protein interaction (PPI) and gene network enrichment analyses via the STRING database, to explore the functional significance of the identified methylation-regulated genes (MRGs). Important pathways enriched among the MRGs include the Wnt signaling pathway, extracellular matrix (ECM) organization, neurogenesis and neuronal differentiation, and maturity-onset diabetes of the young. Zhu et al. [32] reported that the Wnt signaling pathway plays a crucial role in colorectal cancer (CRC), particularly affecting the survival and proliferation of CRC cells, including cancer stem cells. Similarly, Li et al. [33] highlighted that genetic aberrations in components of the Wnt/ -catenin signaling pathway are associated with CRC progression. Karlsson et al. [34] identified the ECM as a potential prognostic marker for CRC due to its critical role within the tumor microenvironment and its possible contribution to metastasis. In agreement, Kim et al. [35] also suggested ECM components as important biomarkers for CRC. Additionally, our PPI network analysis highlighted genes implicated in neurogenesis and neural differentiation. Gut autonomic functions are regulated by the enteric nervous system, and impairments in this system could disrupt interactions with other cellular components, potentially driving CRC tumorigenesis [36,37,38]. Several studies have reported associations between colorectal cancer and Type II diabetes mellitus (T2DM). Liu et al. [39] reviewed evidence demonstrating increased DNA methylation at multiple CpG sites in pancreatic islets of T2DM patients, which significantly reduces PDX1 mRNA expression, impairing insulin secretion. Similarly, Cheng et al. [40] reviewed how insulin resistance might influence tumor growth, thereby linking diabetes and colorectal cancer progression. Survival analysis was carried out for the two spotlight genes, and the high expression of PDX1 was seen to be correlated to low survival, while GNG7 upregulation and downregulation showed similar low survival across samples.
In conclusion, 150 genes were identified as methylation-regulated genes through a comprehensive bioinformatics analysis, suggesting that methylation affects their expression levels. These genes have been associated with a variety of tumors in literature studies, with some specifically linked to colorectal cancer (CRC). We propose the highlighted genes could serve as biomarkers for CRC etiology and disease prognosis. Our study is limited to the secondary analysis, and further experimental tests can further validate the functional insights gained from this study. We look forward to continuing experimental validation as a future direction for this project.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alzahrani S.M. Al Doghaither H.A. Al-Ghafari A.B. General insight into cancer: An overview of colorectal cancer Mol. Clin. Oncol.20211527110.3892/mco.2021.243334790355 PMC 8591689 · doi ↗ · pubmed ↗
- 2Hoang T. Kim H. Kim J. Dietary intake in association with all-cause mortality and colorectal cancer mortality among colorectal cancer survivors: A systematic review and meta-analysis of prospective studies Cancers 202012339110.3390/cancers 1211339133207660 PMC 7697273 · doi ↗ · pubmed ↗
- 3Housini M. Dariya B. Ahmed N. Stevens A. Fiadjoe H. Nagaraju G.P. Basha R. Colorectal cancer: Genetic alterations, novel biomarkers, current therapeutic strategies and clinical trials Gene 202489214785710.1016/j.gene.2023.14785737783294 PMC 12237584 · doi ↗ · pubmed ↗
- 4Ogunwobi O.O. Mahmood F. Akingboye A. Biomarkers in colorectal cancer: Current research and future prospects Int. J. Mol. Sci.202021531110.3390/ijms 2115531132726923 PMC 7432436 · doi ↗ · pubmed ↗
- 5Zygulska A.L. Pierzchalski P. Novel diagnostic biomarkers in colorectal cancer Int. J. Mol. Sci.20222385210.3390/ijms 2302085235055034 PMC 8776048 · doi ↗ · pubmed ↗
- 6Vrba L. Futscher B.W. DNA methylation changes in biomarker loci occur early in cancer progression F 1000 Research 20208210610.12688/f 1000 research.21584.2PMC 699382432047604 · doi ↗ · pubmed ↗
- 7Shen Y. Wang D. Yuan T. Fang H. Zhu C. Qin J. Xu X. Zhang C. Liu J. Zhang Y. Novel DNA methylation biomarkers in stool and blood for early detection of colorectal cancer and precancerous lesions Clin. Epigenet.2023152610.1186/s 13148-023-01443-7PMC 993855336803423 · doi ↗ · pubmed ↗
- 8Zhao G. Liu X. Liu Y. Li H. Ma Y. Li S. Zhu Y. Miao J. Xiong S. Fei S. Aberrant DNA methylation of SEPT 9 and SDC 2 in stool specimens as an integrated biomarker for colorectal cancer early detection Front. Genet.20201164310.3389/fgene.2020.0064332625237 PMC 7314930 · doi ↗ · pubmed ↗