Mesothelin-Associated Anti-Senescence Through P53 in Pancreatic Ductal Adenocarcinoma
Dongliang Liu, Jianming Lu, Changyi Chen, Qizhi Yao

TL;DR
This study shows that the protein mesothelin helps pancreatic cancer cells avoid aging-like states, and targeting it could improve cancer treatments.
Contribution
The study identifies a novel mesothelin-associated anti-senescence mechanism in pancreatic cancer via P53 pathways.
Findings
MSLN deficiency in PDAC cells leads to senescence features like growth arrest and elevated P53 and P21waf1.
MSLN expression inversely correlates with DNA damage/repair pathways in PDAC tissue samples.
IL-8 secretion increases in MSLN-deficient cells, suggesting modulation of the senescence-associated secretory phenotype.
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a deadly disease with limited treatment options. This study focuses on the protein mesothelin (MSLN), which is highly expressed in over 89% of clinical PDAC specimens and helps cancer cells grow and avoid death. This study aims to determine how MSLN prevents cancer cells from entering a state called senescence, where they stop growing. The results show that silencing MSLN gene expression in PDAC cells induced senescence phenotypes, including growth arrest and an increased expression of senescence markers (P53, P21waf1, and P16ink4a), along with higher IL-8 production. Mechanically, high DNA damage/DNA repair activities associated with low MSLN expression may activate the senescence pathway in pancreatic cancer cells. Our findings could lead to better treatment options for pancreatic cancer, potentially improving outcomes for patients.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
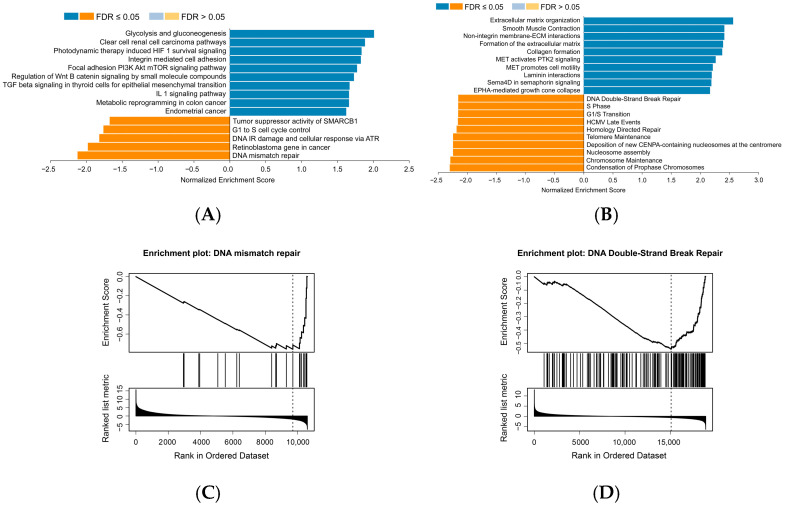 Figure 1
Figure 1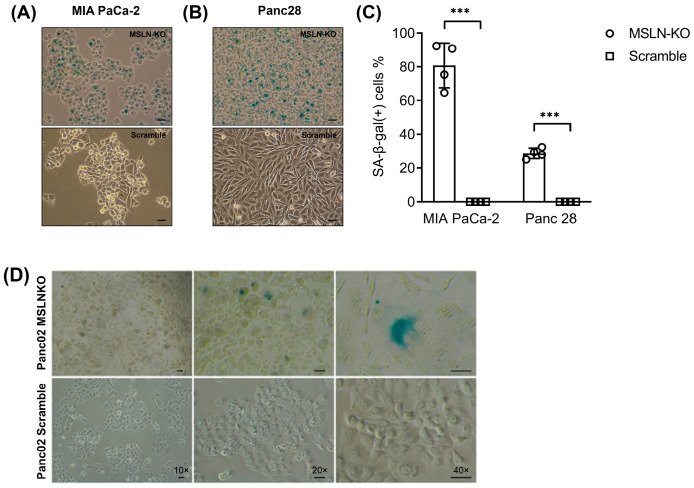 Figure 2
Figure 2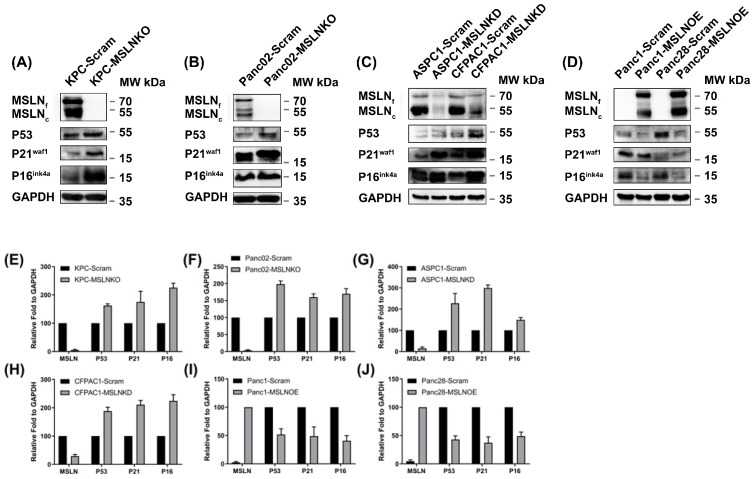 Figure 3
Figure 3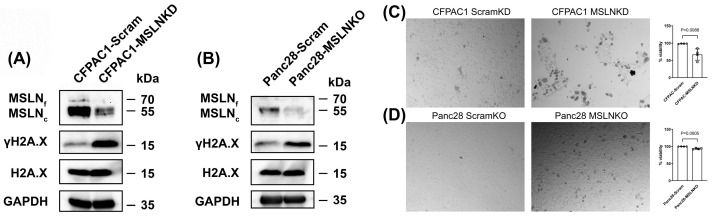 Figure 4
Figure 4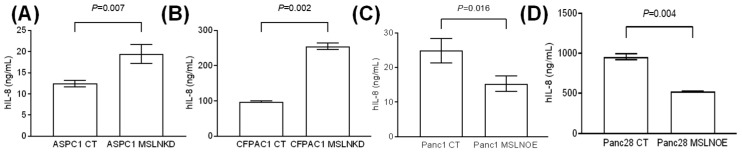 Figure 5
Figure 5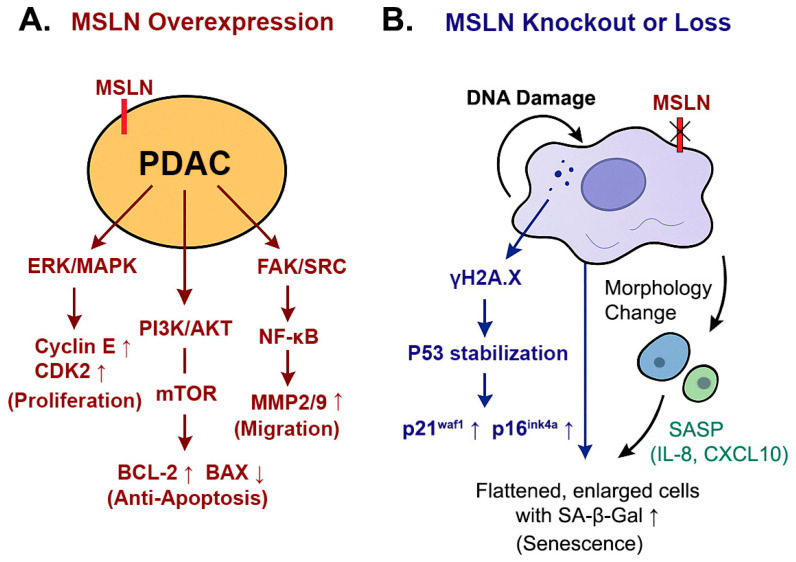 Figure 6
Figure 6- —Dan Duncan Cancer Center Seed Fund from BCM
- —MacDonald Fund from the CHI St. Luke’s Health
- —VA Merit Awards 1
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer Research and Treatments · Occupational and environmental lung diseases · Telomeres, Telomerase, and Senescence
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers and is characterized by an insidious onset, a poor prognosis, and distal metastasis at diagnosis [1]. Due to its late diagnosis and resistance to chemotherapy and immunotherapy, PDAC remains one of the most intractable and devastating malignancies, with a median survival time of merely 6 months post-diagnosis and a 5-year survival rate of less than 7% [2]. Because of its increasing incidence and low cure rate, by 2030, PDAC is projected to exceed liver cancer and become the second leading cause of cancer death for both the male and female sexes in the United States [3]. Deciphering the molecular mechanisms of PDAC’s progression is necessary to design better treatment options.
Cell senescence is considered a growth arrest response to various cellular stresses, including DNA damage, oxidative stress, oncogene activation, etc. [4]. Senescence has been identified as a well-known tumor-suppressive mechanism because the inactivation of senescence effector programs leads to increased tumorigenesis in vivo, indicating that senescence represents an important barrier to the malignant progression of tumors [5]. Senescence-related proteins P53, P21, and P16 are frequently observed to be dysfunctional in many human cancers, which strongly correlates with a worse prognosis [6]. Oncogene-induced senescence (OIS) is crucial for protection against cancer development; it is an anti-proliferative cellular response to a series of oncogenic signaling due to activating mutations of oncogenes or the inactivation of tumor-suppressive genes [7]. The normal ductal epithelium transforming into PDAC undergoes acinar–ductal metaplasia (ADM) and develops into pancreatic intraepithelial neoplasia stages 1 to 3 (PanIN-1 to PanIN-3), with PanIN-3 generally considered to be ductal carcinoma in situ [8]. During the development of PDAC, senescence is reported to be observed only in the ADM and PanIN-1 periods and not in the PDAC stage [9,10]. In a KPC mouse model, pancreatic senescence was observed in PanIN lesions of the pancreas in 6~8-month-old mice, which expressed oncogenic Kras from their endogenous promoters; however, additional oncogenic events or cooperating genetic alterations are needed to break the balance between cellular senescence and proliferation to eventually promote tumor progression [9,10,11].
Mesothelin (MSLN) is a glycoprotein that is highly expressed in PDAC but not in normal pancreas tissues [12,13]. MSLN mediates cellular adhesion by binding to its receptor MUC16 [14,15]. Our previous studies revealed that MSLN acts as a malignant factor in PDAC by promoting cell proliferation and migration and contributing to tumor progression in different mouse models [16,17,18]. However, it is not clear at which point the expression of MSLN emerges during the development of PDAC, while altered MSLN expression has not been shown in the ADM and pancreatic intraepithelial neoplasia stages in immunohistochemical analyses [19]. Notably, MSLN is not expressed in normal pancreas and the ADM and PanIN-1 periods in which senescence can occur, while PDAC without senescence shows aberrantly high MSLN expression [8,9,10]. Although these findings suggest an inverse correlation between MSLN expression and cellular senescence, the specific role of MSLN in regulating senescence in cancer remains unclear.
Hence, the aim of this study was to investigate the potential relationship between MSLN and senescence in the development of PDAC and to obtain insights into the molecular mechanism of MSLN in regulating cellular senescence. This work is the first to demonstrate that MSLN actively suppresses senescence in PDAC cells, representing a novel function beyond its established roles in proliferation and metastasis. By defining a mesothelin-associated anti-senescence (MAAS) effect, our findings reveal a previously unrecognized mechanism of tumor progression and suggest that targeting MSLN could restore senescence, providing a new avenue for therapeutic intervention in PDAC.
2. Materials and Methods
2.1. Cells, Antibodies, and Cell Line Generation
Human PDAC cell lines AsPC1, CFPAC1, MIA Paca2, and Panc1 were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in Yao Lab. Human PDAC cell line Panc28 was obtained from MD Anderson Dr. Craig Logsdon’s lab. Murine PDAC Panc02 cell line was obtained as a gift from Dr. Sabry EL-Naggar, the Medical University of South Carolina [20]. If not specifically stated, all cells were cultured in 1× DMEM (10-017-CM, Corning, Corning, NY, USA ) or 1× RPMI-1640 (10-041-CV, Corning, Corning, NY, USA) medium supplemented with 10% v/v heat-inactivated Fetal Bovine Serum (35-011-CV, Corning, Corning, NY, USA), 100 units/mL penicillin, 100 μg/mL streptomycin, and 250 ng/mL amphotericin B (17-745E, Lonza, Basel, Switzerland). All cell lines used in the study were authenticated with the Short Tandem Repeat (STR) profiling technique by the Cytogenetics and Cell Authentication Core, MD Anderson Cancer Center, University of Texas.
Antibodies anti-MSLN (99966), anti-P53 (13684), anti-P21 (8242), anti-P16 (3033), and anti-H2A.X (7631S) were purchased from Cell Signaling Technology, Danvers, MA, USA; anti-γH2A.X. (AB2893) was purchased from Abcam, Cambridge, UK; and anti-GAPDH (G8795) was obtained from Sigma-Aldrich, St. Louis, MO, USA.
Genetically modified PDAC cell lines with MSLN knockdown, knockout, or overexpression were established as performed previously and maintained by Yao Lab [12,17]. Specifically, CRISPR/Cas9-mediated knockout of MSLN was performed using lentiviral delivery of sgRNA and Cas9. For human PDAC cell lines, lentiviral vectors with sgRNAs targeting MSLN (#HCP290262-LvSG03) and the control (#CCPCTR01-LvSG03) were purchased from GeneCopoeia, Inc., Rockville, MD, USA. Lentivirus was produced in HEK293T cells and used to infect target cells in the presence of polybrene (8 μg/mL). Infected cells were selected with puromycin (5 μg/mL) for 7 days. Following selection, cells were clonally expanded by limiting dilution to establish knockout clones. Gene editing efficiency was confirmed by Sanger sequencing and Tracking of Indels by Decomposition (TIDE) analysis, with >85% of reads showing insertions/deletions (indels) at the target locus across clones. For mouse PDAC cell lines, sgRNAs targeting a murine Msln vector (#MCP301309-LvSG03) and the control vector (#CCPCTR01-LvSG03) were purchased from GeneCopoeia, Inc., Rockville, MD, USA, and delivered via lentiviral transduction. Clonal expansion and validation were performed as described above. MSLN knockdown was achieved using shRNA constructs targeting MSLN (#HSH090262-LVRH1MP) from GeneCopoeia, Inc., Rockville, MD, USA. A scrambled vector (#CSHCTR001-LVRH1MP) was used as a control. Lentivirus was produced in HEK293T cells and used to infect target cells. Stable knockdown was selected using puromycin (5 μg/mL) for 7 days. Knockdown efficiency was confirmed using a Western blot. MSLN overexpression was performed by the lentiviral transduction of full-length human MSLN cDNA cloned into the pLenti6.3/V5™-TOPO™ vector (K531520, Thermo Fisher Scientific Inc., Waltham, MA, USA). Transduced cells were selected using blasticidin (5 μg/mL) for 7 days and validated by Western blot.
2.2. Pathway Enrichment Analysis
To investigate whether MSLN is associated with senescence-related pathways, we analyzed the RNA and protein expression profiles in PDAC patient tissues from the National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium (CPTAC) [21]. The trans-association correlations in PDAC between MSLN and all other top-rated protein coding genes at mRNA and protein levels were analyzed using LinkedOmicsKB (https://kb.linkedomics.org*/*, accessed on 6 March 2025) [22]. A gene set enrichment analysis (GSEA) was performed using the WEB-based GEne SeT AnaLysis Toolkit (WebGestalt, http://www.webgestalt.org/, accessed on 6 March 2025), and the significant genes or proteins from the trans-association results were used as an input list to conduct a pathway enrichment analysis using the KEGG pathway resource [23].
2.3. Senescence-Associated β-Galactosidase Assay (SA-β-Gal) and Cell Viability Assay
The colorimetric SA-β-Gal assay was conducted using the methods described by Fuhrmann-Stroissnigg et al. [24]. Briefly, MSLN gene knockout (MSLN-KO) or scramble PDAC cells were washed three times with cold PBS and then fixed with 2% formaldehyde and 0.2% glutaraldehyde in PBS for 10 min. Following fixation, cells were incubated in SA-β-Gal staining solution (1 mg/mL 5-bromo-4-chloro-3-indolyl-beta-d-galactopyranoside (X-gal), 1× citric acid/sodium phosphate buffer (pH 6.0), 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 150 mM NaCl, and 2 mM MgCl_2_) at 37 °C for 16–18 h. The enzymatic reaction was stopped by washing the cells three times with ice-cold PBS. The cells were counterstained with Hoechst solution and analyzed with a fluorescence microscope.
Cell viability was assessed directly in 24-well plates using the 0.2% Trypan Blue exclusion assay. Briefly, the cells were gently washed with phosphate-buffered saline (PBS) to eliminate residual serum. Subsequently, 200 µL of 0.2% Trypan Blue solution was added to each well, and the mixture was incubated at room temperature for 5 min. After incubation, the excess dye was gently aspirated, and the cells were immediately visualized under an inverted light microscope. Viable cells appeared clear, while non-viable cells were stained blue. The numbers of stained (non-viable) and unstained (viable) cells were counted from representative fields in each well.
The quantification of SA-β-gal-positive cells or Trypan Blue-stained cells was conducted using ImageJ software (version 1.53t, National Institutes of Health, Bethesda, MD, USA) [25]. The brightfield images were converted to 8-bit grayscale and analyzed using the Color Deconvolution plugin to distinguish positively stained (blue) cells from the background. Positive cells were counted using the Cell Counter plugin or the Analyze Particles function after segmentation. The percentage of SA-β-gal-positive cells was calculated by dividing the number of blue-stained cells by the total number of cells per field. For the Trypan Blue exclusion assay, the percentage of viable cells was calculated by dividing the number of unstained (viable) cells by the total number of cells per field. A minimum of three randomly selected fields and three independent biological replicates were analyzed for each condition.
2.4. Western Blot Assay
Protein purification and a Western blot assay were carried out as previously described [12]. Cell pellets were collected and lysed using 1× RIPA Lysis Buffer (20-188, Millipore Sigma, Burlington, MA, USA) supplemented with 1× Protease and Phosphatase Inhibitor Cocktail (78440, Thermo Scientific, Waltham, MA, USA); following lysis, all cell lysates were centrifuged at 12,000 rpm for 15 min at 4 °C, and the protein concentration of supernatant was determined by using a PierceTM BCA Protein Assay Kit (23227, Thermo Scientific, Waltham, MA, USA). A Western blot assay was used to detect MSLN, while senescent markers and DNA damage response markers were determined with antibodies anti-MSLN (99966), anti-P53 (13684), anti-P21 (8242), anti-P16 (3033), anti-H2A.X (7631S) (Cell Signaling Technology, Danvers, MA, USA), anti-γH2A.X. (AB2893, Abcam, Cambridge, UK), and anti-GAPDH (G8795, Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions. Immunoreactivity protein complexes were detected by using horseradish peroxidase (HRP)-conjugated anti-rabbit (7074) or anti-mouse IgG antibodies (7076) (Cell Signaling Technology, Danvers, MA, USA) and SuperSignal™ West Pico PLUS Chemiluminescent Substrate (34580, Thermo Scientific, Waltham, MA, USA). For blot quantification, the Gel Analysis tool in ImageJ was used [25]. Bands were selected using the rectangular selection tool, and intensity profiles were plotted to measure the area under the curve for each band. Band intensities were normalized to loading control, and the results were expressed as relative expression levels. Quantification was based on triplicate biological replicates.
2.5. Enzyme-Linked Immunosorbent Assay (ELISA)
The chemokine IL-8/CXCL-8, a very common senescence-associated secretory phenotype overexpressed by most senescent cells [26], was measured by using the LEGEND MAX™ Human IL-8 ELISA Kit (431507, BioLegend, San Diego, CA, USA) according to the manufacturer’s instructions. The optical density values were measured using a microplate reader at 450 and 570 nm, and values at 570 nm were subtracted from the absorbance at 450 nm for subsequent data analysis.
2.6. Statistical Analysis
All quantifications from the SA-β-gal staining and Trypan Blue staining assays reflect at least three samples with at least 100 events counted (typically in three different areas) each. Unless stated, the data are presented as arithmetic means ± standard deviation (s.d.), and statistical analyses were conducted based on unpaired two-sided t-tests. p < 0.05 was considered statistically significant. Data analyses were performed by using GraphPad Prism Version 8.4 [27].
3. Results
3.1. MSLN Negatively Correlates with Cell Senescence-Related Signaling Pathways in Human PDAC Samples
The CPTAC is a national effort to accelerate the understanding of the molecular basis of cancer through the application of large-scale proteome and genome analyses or proteogenomics [28]. A total of 105 tissue samples from PDAC patients and 44 normal tissue samples from the CPTAC were retrieved to conduct a GSEA in this study. As a result of the WikiPathways cancer analysis (FDR < 0.05 and 1000 permutations), 10 positive related categories and 5 negative related categories were identified as enriched categories, as shown in Figure 1A. The GSEA revealed significant enrichment of MSLN trans-association genes in the pathways of tumor suppressor activity, cell cycle control, DNA damage response, and mismatch repair; the rank plot demonstrated a skewed distribution of these genes toward the bottom of the ranked list, suggesting that MSLN may participate in those biological processes by negatively regulating corresponding pathways. In addition, based on the Reactome pathway analysis (FDR < 0.05 and 1000 permutations), 33 positive related categories and 203 negative related categories were identified as enriched categories, and 10 of the most significant categories and representatives in the reduced sets are shown in Figure 1B. Similarly, the cell cycle-related pathway and DNA repair pathway are significantly enriched in the top MSLN-negative correlation categories. Reactome and WikiPathways are two of the most popular freely available databases for biological pathways [29,30]. Interestingly, both database analyses showed that component genes mainly enriched in cell cycle pathways and DNA damage/DNA repair-related pathways with negative normalized enrichment score (NES) values were negatively correlated with MSLN expression. This suggests that high MSLN expression in PDAC may be associated with the repression or reduced activity of these biological pathways (Figure 1A–D), which is consistent with other reports showing that the senescence pathway is being triggered by a variety of stimuli, including DNA damage, mitochondrial dysfunction, and tumor suppressor loss [31,32,33]. Therefore, we hypothesized that inhibiting the high expression of MSLN in PDAC may induce significant DNA damage/DNA repair activities and thereby activate the senescence pathway.
3.2. MSLN Loss Leads to Morphological Changes and Cell Senescence in PDAC Cells
To determine whether MSLN is associated with anti-senescence, we genetically modified MSLN expression levels in both mouse and human PDAC cell lines and determined the changes in these cells when compared with the control cells. Senescence-associated beta-galactosidase (SA-β-gal) is a hydrolase enzyme only found in senescent cells that can catalyze the hydrolysis of β-galactosides into monosaccharides [34]. SA-β-gal activity was strongly associated with senescent cells since it was not detectable in quiescent cells or terminally differentiated cells; therefore, it is now a widely used biomarker in studies of cellular senescence in vitro and in vivo [35]. Here, an SA-β-gal activity assay was performed to distinguish senescent cells from non-senescent cells. We utilized CRISPR/Cas9 technology to target the DNA site (5′-CAACGGCTCGACCCCTGTTG-3′) of the MSLN gene to deplete its expression in the PDAC cell lines MIA PaCa-2 and Panc28, and the MSLN knockout (MSLN-KO) was subsequently confirmed using PCR-based DNA sequencing (Supplementary Figure S1). SA-β-gal staining showed that the proportion of SA-β-gal-positive cells increases from 0% in the cultures of CRISPR/Cas9 scramble cells to 81% in MIA PaCa-2 MSLN-KO cells (Figure 2A) and to 29% in Panc 28 MSLN-KO cells (Figure 2B). Figure 2C shows the quantification of SA-β-gal-positive cells in MSLN-KO cells vs. scramble control cells. Similarly, we found that MSLN-KO in mouse Panc02 cells also show a significant cellular senescence phenotype (with more SA-β-gal staining, cells become flattened and larger) when compared with the control cells (Figure 2D).
3.3. MSLN Is Negatively Correlated with Senescence Regulators
P53, p21^waf1^, and p16^INK4a^ are major cell cycle regulators that control cellular senescence [36,37]. Therefore, we further studied the expression levels of these three cellular senescence regulators in PDAC cells before and after MSLN genetic modification (knockout, knockdown, or overexpression). As shown in Figure 3, we found that compared with scramble control cells, MSLN-KO or MSLN-KD cells showed elevated expressions of P53, p21^waf1^, and p16^INK4a^, whereas MSLN-OE PDAC cells showed decreased senescence regulator expression (Figure 3A,B: two PDAC mouse cell lines; Figure 3C,D: two PDAC human cell lines). For the mouse PDAC cells, we observed higher fundamental MSLN expression in KPC-Scram than in Panc02-Scram cells; the senescence regulators P53, p21^waf1^, and p16^INK4a^ were dramatically upregulated after MSLN knockout (Figure 3A,B). Similarly, in the human PDAC cell lines, higher expressions of senescence regulators P53, p21^waf1^, and p16^INK4a^ were detected with MSLN knockdown (Figure 3C). The overexpression of MSLN in Panc1 and Panc28 PDAC cells showed reduced expressions of P53, p21^waf1^, and p16^INK4a^ proteins (Figure 3D). The expression levels of MSLN, P53, P21, and P16 in different cells shown in the Western blot were quantified by using ImageJ software (Figure 3E–J). Our findings indicate that MSLN may suppress cellular senescence activation by inhibiting the expression of major cell cycle regulators in PDAC cells.
3.4. MSLN Deficiency Induces DNA Damage and Reduces Cell Viability in PDAC Cells
To further verify whether the senescence caused by MSLN deficiency involves DNA damage in PDAC cells, we examined the level of γH2A.X, which is phosphorylated H2A.X and a well-established marker of DNA damage that plays a key role in the repair of DNA double-strand breaks. A Western blot analysis revealed a substantial increase in γH2A.X expression in CFPAC1-MSLNKD and Panc28-MSLNKO cells compared to their respective scrambled controls (Figure 4A,B). This increase was not accompanied with changes in the total H2A.X level, indicating that the observed upregulation of γH2A.X occurred due to phosphorylation events rather than changes in overall histone abundance. These findings suggest that the depletion of MSLN activates DNA damage signaling pathways in PDAC cells. We further evaluated the impact of MSLN deficiency on cell viability by using a Trypan Blue exclusion assay. As shown in Figure 4C, CFPAC1 cells with MSLN knockdown (CFPAC1-MSLNKD) exhibited a larger number of non-viable (Trypan Blue-positive) cells compared to the scrambled control (CFPAC1-Scram), indicating increased cell death. A quantitative analysis revealed a significant reduction in cell viability in the MSLNKD group, with viability decreasing to 67% compared to near-complete viability (98.4%) in the control group (p = 0.0088). Similarly, Panc28 cells with CRISPR-mediated MSLN knockout (Panc28-MSLNKO) showed a moderate but significant reduction in cell viability relative to Panc28-Scram controls (Figure 4D). Together, these data suggest that the depletion of MSLN compromises cell viability in PDAC cells, potentially through mechanisms associated with increased cellular senescence.
3.5. MSLN Suppresses the Production of the Senescence-Associated Secretory Phenotype
Senescent cells have been reported to experience a series of changes before and after, such as morphology change, and the secretion of pro-inflammatory factors termed the senescence-associated secretory phenotype (SASP) [38,39]. SASP-related factors include pro-inflammatory cytokines (such as IL-6 and IL-8) and chemokines (such as CXCL-1/3 and CXCL-10) [40]. Our ELISA data show that MSLN-KD cells showed a significant increase in IL-8 production that was secreted into the cell culture supernatant compared with the control cells (Figure 5A: ASPC1 cells; Figure 5B: CFPAC1 cells), while the MSLN-OE PDAC cells showed obviously downregulated IL-8 levels in the cell culture supernatant (Figure 5C: Panc1 cells; Figure 5D: Panc28 cells). As shown in Figure 5A,B, IL-8 production in ASPC1 or CFPAC1 MSLN-KD cells was significantly greater than in scramble ASPC1 or CFPAC1 cells (19.3 ng/mL vs. 12.4 ng/mL, p = 0.007 and 254.7 ng/mL vs. 98.1 ng/mL, p = 0.002, respectively). Similarly, in Figure 5C,D, IL-8 production by Panc1 or Panc28 MSLN-OE cells was significantly lower than in Panc1 or Panc28 control cells (15.2 ng/mL vs. 24.7 ng/mL, p = 0.016 and 525.0 ng/mL vs. 956.2 ng/mL, p = 0.004, respectively).
4. Discussion
Our previous studies demonstrated that mesothelin (MSLN) plays a significant role in pancreatic ductal adenocarcinoma (PDAC) by promoting cell proliferation, migration, and invasion while inhibiting apoptosis [12,13,16,17]. Importantly, this study presents novel evidence that MSLN inhibits cellular senescence—a previously underexplored mechanism contributing to its oncogenic role. Our findings align with data from the CPTAC, which show that high MSLN expression in human PDAC is associated with reduced activity in DNA damage response, repair pathways, and cell cycle control mechanisms. This suggests that MSLN helps PDAC cells evade senescence by downregulating these critical pathways, thereby promoting tumor progression. Using CRISPR/Cas9 to knock out MSLN in PDAC cell lines, we found that MSLN-KO cells exhibited hallmark features of cellular senescence, such as growth arrest, morphological changes, and increased senescence-associated β-galactosidase (SA-β-gal) staining. Additionally, we observed elevated levels of senescence-associated molecules (P53, P21^waf1^, and P16^ink4a^) and the pro-inflammatory cytokine IL-8 in MSLN-KO cells. These findings highlight the role of MSLN in preventing cellular senescence in PDAC, suggesting that targeting MSLN may offer a novel approach to enhance the effectiveness of PDAC therapies.
There are limited studies on the role of MSLN in cellular senescence. In our previous study, we examined the effect of MSLN on pancreatic cancer cell proliferation, cell cycle progression, the expression of cell cycle regulatory proteins, and signal transduction pathways in different PDAC cell lines [16,17,18]. Our data show that MSLN-overexpressed PDAC cells enhanced proliferation more than control cells; however, MSLN gene knockdown decreased cell proliferation and postponed the progression of cell entry into the S phase compared with the control cells [13]. Another study indicated that MSLN-overexpressed cells upregulated the expressions of cyclin-dependent kinase 2 (CDK2) and cyclin E, which correlated with significantly increased cell proliferation and faster cell cycle progression [12]. The cyclin E/Cdk2 complex is believed to play a key role in the regulation of the cell cycle [41]. Morisaki et al. demonstrated an impaired activation of this complex in senescent cells [42]. Similarly, Yoshida and colleagues showed that CSN5 can control premature senescence by depending on the CDK2/cyclin E axis in human diploid fibroblasts [43]. In addition, CDK2 plays a significant role in suppressing oncogene-induced senescence since CDK2 suppression-induced senescence was detected in various human cancer cell types [44,45]. In another study on ovarian cancer, Li et al. revealed that MSLN displayed the most negative association with several significant KEGG pathways, which were mainly related to the p53 signaling pathway, cellular senescence, and cell cycle [46], but no more data about the details of the MSLN–senescence relationship were provided in their study. The P38 MAPK signaling pathway was reported to be activated in cellular senescence and stress-induced premature senescence triggered by oncogene and culture shock [47,48]; however, there is no evidence that the P38 pathway is significantly altered upon the inhibition of MSLN expression. Therefore, MSLN-induced senescence inhibition may be mainly associated with other signaling responses instead of the P38 MAPK pathway.
In the present study, we provided primary evidence that MSLN inhibits senescence in PDAC cell lines. For the first time, our findings revealed that MSLN-associated anti-senescence (MAAS) is accompanied by elevated levels of key senescence regulators, including P53, P21^waf1^, and P16^ink4a^. These proteins play crucial roles in the cellular response to stress, with P53 acting as a transcription factor that can induce cell cycle arrest, apoptosis, or senescence in response to DNA damage [36]. P21, a downstream target of P53, inhibits cyclin-dependent kinases, leading to cell cycle arrest [49]. P16, another important regulator, inhibits CDK4/6, preventing the phosphorylation of RB and thereby enforcing cell cycle arrest [50]. In addition, the fact that MSLN-deficient cells led to a moderate but significant reduction in cell viability further supports the finding that MSLN loss suppresses tumor cell proliferation by inducing a senescent-like state rather than causing overt cytotoxicity. This reduction reflected a shift toward a non-proliferative state consistent with senescence rather than widespread cell death. We also observed increased γH2AX levels in MSLN-deficient cells, indicating the activation of the DNA damage response and supporting the role of MSLN loss in triggering cellular stress and senescence. Together, these findings reinforce the notion that MSLN is critical for maintaining proliferative capacity and genomic stability in pancreatic cancer cells. A schematic model of this MSLN-mediated anti-senescence mechanism is shown in Figure 6.
Our study shows that PDAC senescent cells display an enlarged, flattened cell shape and elevated senescence-associated β-galactosidase activity, a gold standard for identifying senescent cells. Although the component of SASP is very complicated, recent studies demonstrated that numerous unique proteins with immunomodulatory properties are secreted from senescent cells, including inflammatory cytokines, chemokines, growth factors, and matrix metalloproteinases [5]. In the tumor microenvironment, for example, IL-8, IL-12, and tumor necrosis factor (TNF) can recruit immune cells such as M1-like macrophages, enhancing the removal of detrimental senescent and aging cells that are consistent with the tumor suppression effect of senescence [51,52]. To detect the SASP level in cell culture supernatants from senescent PDAC cells induced by MSLN knockdown or overexpression, we performed an IL-8 ELISA. Our data clearly show significantly elevated IL-8 secretion in MSLN knockdown PDAC cell lines compared to the control cells; on the contrary, IL-8 secretion dramatically decreased in MSLN-overexpressed PDAC cells. These findings suggest that the MSLN expression level is inversely correlated with the proportion of PDAC cells in the senescent state, which is consistent with our SA-β-gal data. As another key factor of the SASP, IL-6 can not only promote tumorigenesis and cell proliferation but also exert tumor-suppressive functions depending on the cellular context [5]. Our previous investigation indicated that MSLN-activated NF-kB induces upregulated IL-6 expression, which acts as a growth factor to support PDAC cell proliferation and anti-apoptosis through a novel auto/paracrine IL-6/sIL-6R trans-signaling pathway [17]. Therefore, we hypothesized that the high-MSLN-induced upregulated IL-6 axis (MSLN^H^IL6^H^) is an example of major survival/proliferation signaling in supporting PDAC cell growth; on the contrary, low-MSLN or MSLN-inhibition-induced upregulated IL-6 secretion (MSLN^L^IL6^H^) from senescent cells may play an important role in suppressing tumor progression. More studies are needed to verify this hypothesis.
MSLN knockout studies provide strong mechanistic insights, allowing for these findings to be translated into clinical therapy, which requires a deeper exploration of potential pharmacologic strategies to inhibit MSLN. Currently, several monoclonal antibodies (e.g., amatuximab) and antibody–drug conjugates (e.g., anetumab ravtansine) targeting MSLN are being investigated in preclinical and clinical trials, particularly for mesothelioma [53,54] and ovarian cancer [55,56]. These agents could potentially be repurposed or tested in PDAC settings to evaluate whether pharmacological MSLN inhibition induces tumor senescence and improves therapy response. Furthermore, reported compounds such as synthetic inhibitors have shown promise in modulating mesothelin expression [57], though their mechanisms in the context of senescence remain to be clarified. Another important translational direction involves targeted delivery. Nanoparticle-mediated drug delivery systems or ligand-targeted approaches (e.g., using MUC16-binding peptides) could enhance the selective inhibition of MSLN at the tumor site, minimizing off-target effects [58,59].
Recent progress in senolytic therapies has highlighted their potential in selectively eliminating senescent cells to mitigate age-related diseases and improve cancer treatment outcomes [60,61,62]. Additionally, combination strategies involving senolytics and chemotherapeutic agents have shown promise in reducing therapy-induced senescence and tumor relapse [63,64]. Ongoing clinical trials, including those evaluating dasatinib and quercetin (D + Q) in fibrotic and metabolic diseases, are paving the way for the translational application of senolytics across a range of senescence-associated pathologies, including cancer and degenerative disorders [60,65,66].
This study opens the possibility of combining MSLN-targeted agents with senolytic agents to synergistically induce cancer cell death. Targeting MSLN could therefore be a promising strategy to induce senescence in PDAC cells and improve therapeutic outcomes for PDAC patients. In the future, we will explore a combination strategy using an MSLN blockade in conjunction with senolytics (e.g., dasatinib and quercetin or navitoclax) in PDAC mouse models. While MSLN inhibition is expected to induce a senescent phenotype in PDAC cells via the activation of the P53/P21 and P16/RB pathways, senolytics could be used to selectively clear these senescent tumor cells, potentially reducing the pro-tumorigenic effects of chronic SASP secretion (e.g., IL-6 and IL-8). This sequential treatment strategy may enhance therapeutic efficacy by inducing tumor cell cycle arrest and then eliminating residual senescent cells.
5. Conclusions
This study identifies a novel function of MSLN as a key suppressor of cellular senescence in PDAC, expanding its known oncogenic profile beyond proliferation and survival promotion. By modulating MSLN expression in human and murine PDAC models, we demonstrate that MSLN deficiency triggers classical senescence hallmarks—including growth arrest, morphological changes, the activation of the P53/P21^waf1^ and P16^ink4a^ pathways, elevated γH2AX levels, and the increased secretion of SASP factor IL-8—while preserving overall cell viability. These findings define a mesothelin-associated anti-senescence (MAAS) mechanism that enables PDAC cells to evade stress-induced growth arrest and sustain malignant progression. Importantly, our results provide compelling evidence that MSLN acts as a molecular brake on senescence, positioning it as a promising therapeutic target. Disrupting MAAS may not only restore tumor-suppressive senescence programs but also sensitize PDAC cells to senolytic therapies, offering a novel translational strategy to enhance treatment responsiveness and improve clinical outcomes in this aggressive cancer type.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Wood L.D. Canto M.I. Jaffee E.M. Simeone D.M. Pancreatic cancer: Pathogenesis, screening, diagnosis, and treatment Gastroenterology 202216338640210.1053/j.gastro.2022.03.05635398344 PMC 9516440 · doi ↗ · pubmed ↗
- 2Fan M. Deng G. Ma Y. Si H. Wang Z. Dai G. Survival outcome of different treatment sequences in patients with locally advanced and metastatic pancreatic cancer BMC Cancer 2024246710.1186/s 12885-024-11823-838216928 PMC 10785544 · doi ↗ · pubmed ↗
- 3Rahib L. Wehner M.R. Matrisian L.M. Nead K.T. Estimated projection of US cancer incidence and death to 2040 JAMA Netw. Open 20214 e 21470810.1001/jamanetworkopen.2021.470833825840 PMC 8027914 · doi ↗ · pubmed ↗
- 4Kumari R. Jat P. Mechanisms of cellular senescence: Cell cycle arrest and senescence associated secretory phenotype Front. Cell Dev. Biol.2021964559310.3389/fcell.2021.64559333855023 PMC 8039141 · doi ↗ · pubmed ↗
- 5Dong Z. Luo Y. Yuan Z. Tian Y. Jin T. Xu F. Cellular senescence and SASP in tumor progression and therapeutic opportunities Mol. Cancer 20242318110.1186/s 12943-024-02096-739217404 PMC 11365203 · doi ↗ · pubmed ↗
- 6Yamauchi S. Takahashi A. Cellular senescence: Mechanisms and relevance to cancer and aging J. Biochem.202517716316910.1093/jb/mvae 07939551937 PMC 11879292 · doi ↗ · pubmed ↗
- 7Bartkova J. Rezaei N. Liontos M. Karakaidos P. Kletsas D. Issaeva N. Vassiliou L.V.F. Kolettas E. Niforou K. Zoumpourlis V.C. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints Nature 200644463363710.1038/nature 0526817136093 · doi ↗ · pubmed ↗
- 8Marstrand-DaucéL. Lorenzo D. Chassac A. Nicole P. Couvelard A. Haumaitre C. Acinar-to-ductal metaplasia (ADM): On the road to pancreatic intraepithelial neoplasia (Pan IN) and pancreatic cancer Int. J. Mol. Sci.202324994610.3390/ijms 2412994637373094 PMC 10298625 · doi ↗ · pubmed ↗