Multi-Transcriptomic Analysis Reveals GSC-Driven MES-Like Differentiation via EMT in GBM Cell–Cell Communication
Weichi Wu, Po Zhang, Dongsheng Li, Kejun He

TL;DR
This study shows that glioma stem cells in brain tumors communicate in a way that promotes aggressive tumor growth through a process called epithelial-mesenchymal transition.
Contribution
The study reveals a novel link between glioma stem cell communication and epithelial-mesenchymal transition (EMT) in driving tumor progression.
Findings
GSC communication patterns are strongly associated with epithelial-mesenchymal transition (EMT).
EMT-related genes are highly expressed in mesenchymal-like GSCs and linked to poor patient prognosis.
MES-like GSCs are more aggressive and associated with worse clinical outcomes than other GSC subtypes.
Abstract
Background: Glioblastoma (GBM) is the most malignant brain tumor, with a cellular hierarchy dominated by glioma stem cells (GSCs). Understanding global communications among GSCs and other cells helps us identify potential new therapeutic targets. In this study, multi-transcriptomic analysis was utilized to explore the communication pattern of GSCs in GBM. Methods: CellChat was used to quantitatively infer and analyze intercellular communication networks from GBM single-cell RNA-sequencing (scRNA-seq) data. Gene set enrichment analysis (GSEA) was conducted to identify specific biological pathways (epithelial–mesenchymal transition, EMT) involved in the communication pattern of GSCs. Spatial transcriptomic database was used to support the relationship between EMT and GSC proliferation. Single-sample GSEA (ssGSEA) was employed to assess which GSC state exhibited the strongest association…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
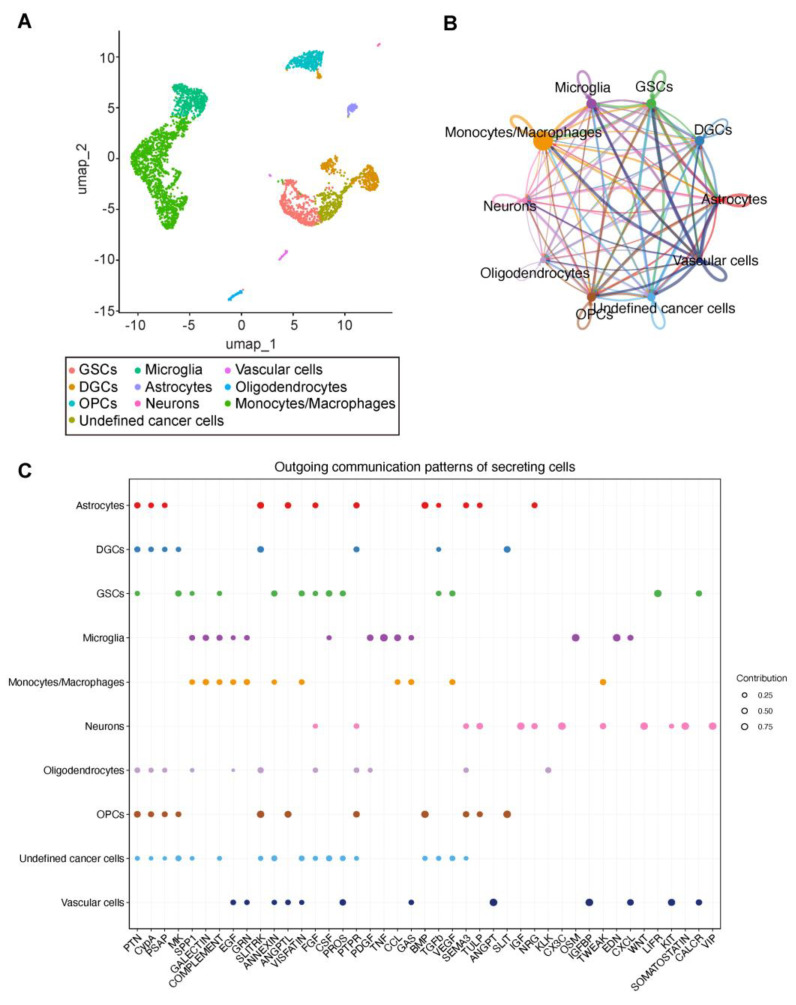 Figure 1
Figure 1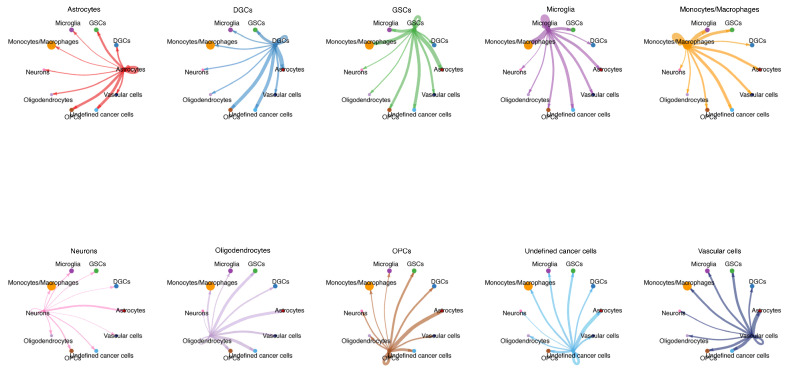 Figure 2
Figure 2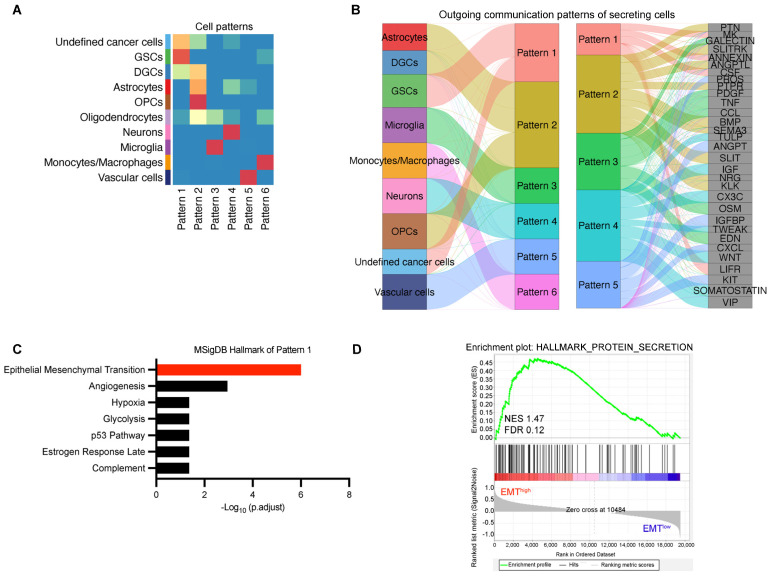 Figure 3
Figure 3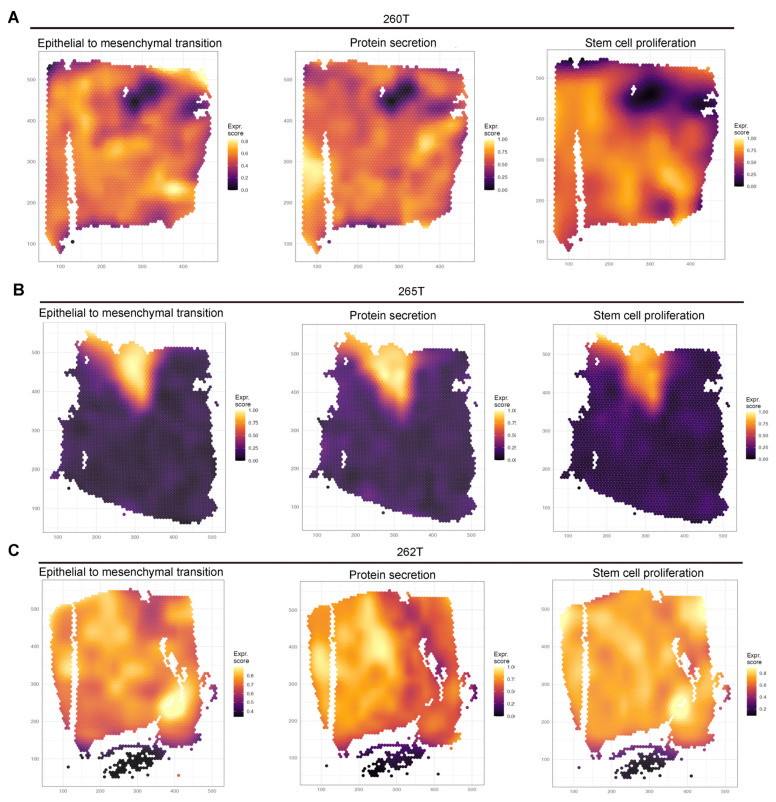 Figure 4
Figure 4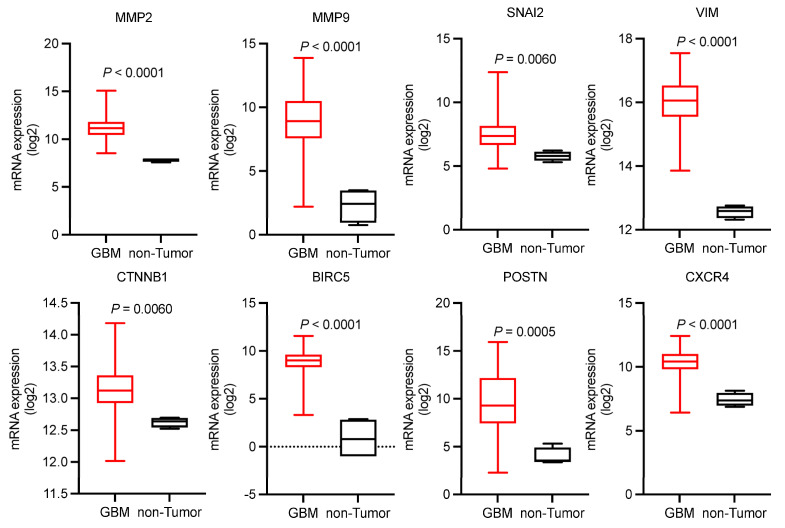 Figure 5
Figure 5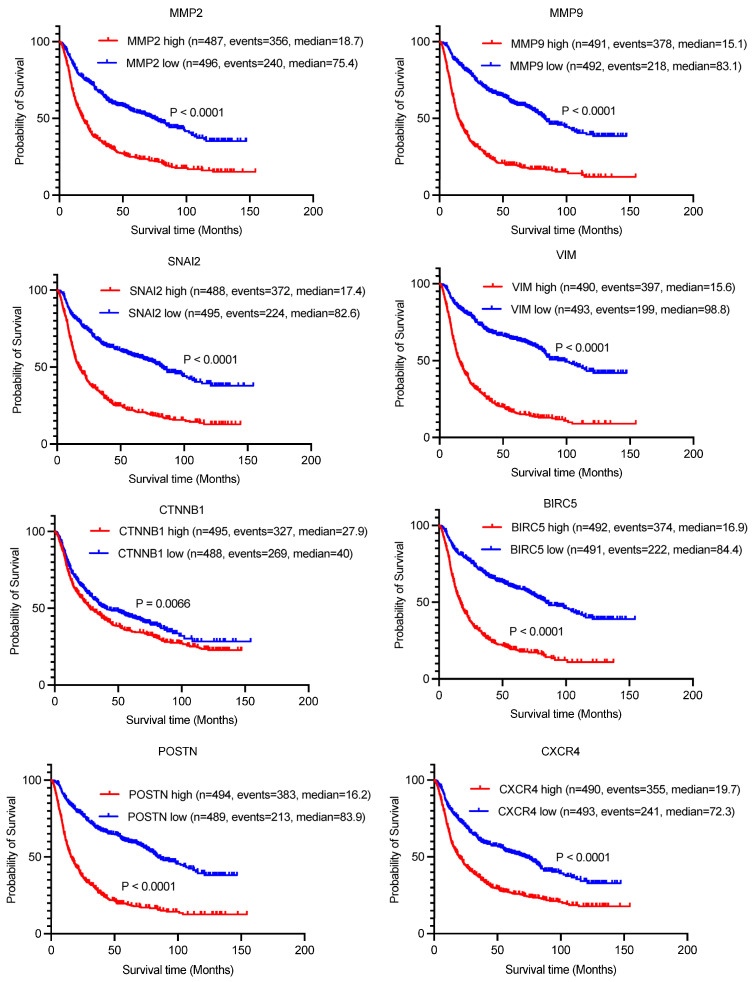 Figure 6
Figure 6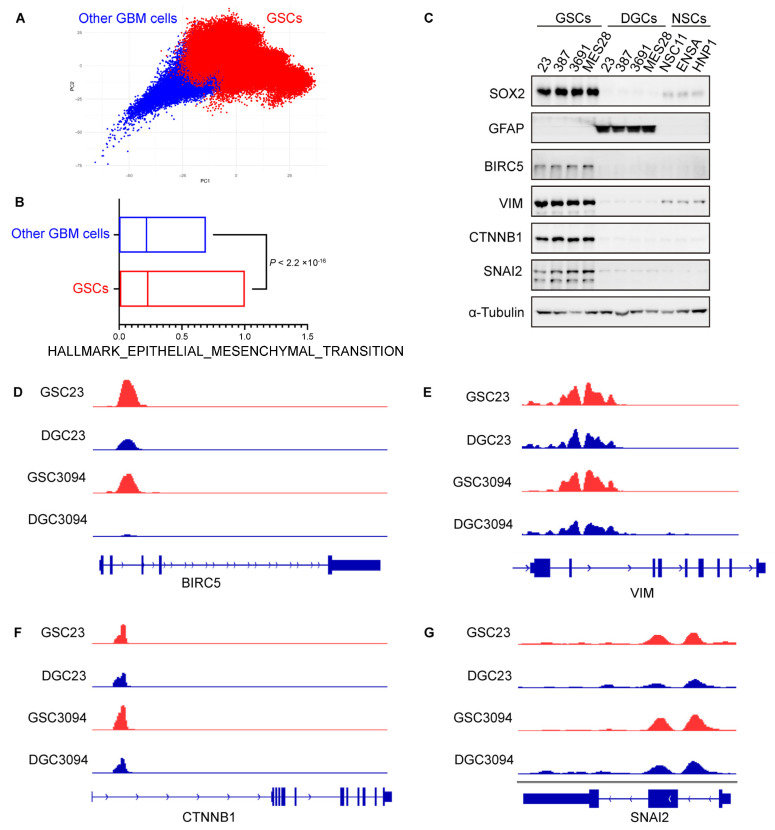 Figure 7
Figure 7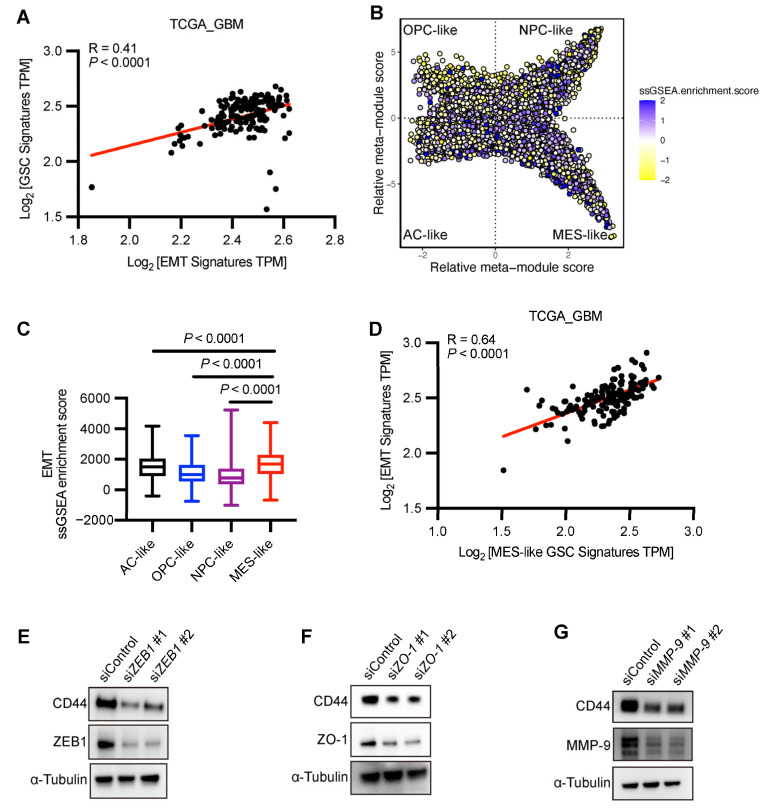 Figure 8
Figure 8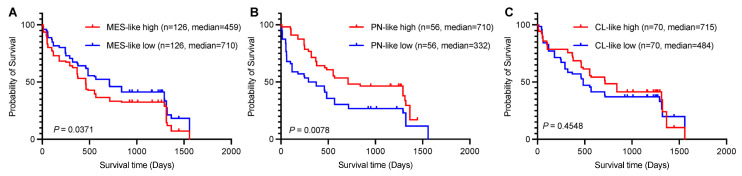 Figure 9
Figure 9- —National Natural Scientific Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicroRNA in disease regulation · Advanced biosensing and bioanalysis techniques · Single-cell and spatial transcriptomics
1. Background
Glioblastoma (GBM) is the predominant malignant and most lethal tumor of the central nervous system (CNS), carrying the highest mortality rates [1]. Epidemiological studies indicate that the incidence rate of GBM is approximately 5.26/100,000, with nearly 17,000 new diagnoses annually [2,3,4]. The current standard of care (SoC) for GBM, including maximal surgical resection with preservation of neurologic function, chemoradiation, and adjuvant chemotherapy [5], offers only limited survival benefit, and almost all patients eventually recur, in part due to the presence of glioma stem cells (GSCs) [6]. As at the apex of cellular hierarchies in GBM, the self-renewing GSCs contribute to neovascularization, immunosuppressive adaptation, infiltrative growth patterns, and multidrug resistance phenotypes during tumor progression [7,8,9,10]. Therapeutic targeting of GSCs holds significant promise for GBM management. However, developing effective GSC-targeting strategies remains challenging due to their heterogeneity, plasticity, and varied therapeutic responses [11,12,13].
Recent studies have established foundational insights into GSC heterogeneity and epithelial–mesenchymal transition (EMT). For instance, Neftel et al. [14] delineated four GSC subtypes with distinct transcriptional programs, including oligodendrocyte precursor cell-like (OPC-like), neural progenitor cell-like (NPC-like), astrocyte-like (AC-like), and mesenchymal-like (MES-like), highlighting microenvironment-driven plasticity. However, the dynamic interplay between subtype-specific transcriptional regulators and EMT effectors remains unresolved.
Cells communicate through released chemicals and surface signals to guide key decisions like starting growth, triggering self-destruct, moving locations, or developing into specific cell types [15,16,17]. The tumor microenvironment (TME) in GBM is highly complex, containing various cell types and extracellular components. Understanding how tumor cells, particularly GSCs, interact with surrounding cells could reveal key mechanisms driving GBM progression and help identify potential treatment targets. The interplay between GSCs and other GBM cells within TME is multi-dimensional, with biophysical properties integral to oncology, and proposed as a source for new strategies for GBM management.
Single-cell RNA sequencing (scRNA-seq) allows researchers to study cell differences and development patterns in great detail [18,19]. Several strategies have been recently applied to infer cell–cell communication from scRNA-seq data [20,21,22,23,24,25,26], such as CellChat [20], Single-CellSignalR [22], and NicheNet [25]. Other than scRNA-seq, recent progress in spatially resolved transcriptomic methods allows researchers to study how cells are arranged spatially in tissues [27]. Combining these spatial data with scRNA-seq results could help provide a better understanding of cellular crosstalk in GBM [28].
Therefore, our study aims to identify communication patterns of GSCs in GBM and their impact on disease progression. Through integrated analysis of multiple scRNA-seq datasets and spatial transcriptomic data, we found that GSC communication patterns are closely associated with EMT. Further analysis showed that EMT may promote increased proportions of MES-like subtypes in GSCs, which are strongly linked to poorer patient outcomes. Importantly, targeting EMT-related genes in GBM could serve as a potential therapeutic strategy for future treatment development.
2. Materials and Methods
2.1. Data Source and Processing
Data were obtained from the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/, accessed on 16 March 2025) database at the National Center for Biotechnology Information (NCBI) in the United States. The information on the datasets is presented in Table 1.
For the GSE84465 dataset, following data normalization, we performed differential expression analysis across all cells, conducted principal component analysis (PCA), and subsequently applied clustering and UMAP dimensionality reduction. Cell annotation was performed using markers for GSCs and differentiated glioma cells (DGCs) defined by Martina et al. [29], while employing the CellMarker database (http://xteam.xbio.top/CellMarker/#, accessed on 16 March 2025) as a reference for other cell types.
For the GSE129438 dataset, the BW-format files were visualized using the Integrative Genomics Viewer (IGV) software (version 2.17.0), with genomic data aligned to the human reference genome assembly (GRCh37/hg19).
For the GSE131928 dataset, the pre-processed data provided by the authors through the GEO repository were directly employed for subsequent analytical procedures.
2.2. Cell–Cell Interaction Analysis
The R package CellChat V.1.6.114 was used to quantitatively infer and analyze intercellular communication networks using scRNA-seq data of GSE84465. CellChat employs a computational framework integrating network topology analysis and pattern recognition algorithms to systematically infer principal signaling input–output relationships among cellular populations.
Inference of intercellular communications: identification of differentially expressed signaling genes. To infer the cell state-specific communications, we first identified differentially expressed signaling genes across all cell groups within the scRNA-seq dataset using the Wilcoxon rank sum test with a significance level of 0.05.
2.3. Functional Enrichment Analysis
The clusterProfiler package (v 3.14.3) was used to perform Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses on the cellular communication pattern genes of GSCs. The gene set enrichment analysis (GSEA) software (v 4.3.3) was obtained from the GSEA website (http://software.broadinstitute.org/gsea/index.jsp, accessed on 17 March 2025) and used for GSEA enrichment analysis based on the MSigDB database [30,31] (http://www.gsea-msigdb.org/gsea/downloads.jsp, accessed on 17 March 2025). Additionally, single-sample GSEA (ssGSEA) was performed to quantify the EMT-related enrichment score of various GSC subtypes within GBM.
2.4. Processing of GBM Spatial Transcriptome Sequencing Data
GBM spatial transcriptome sequencing data were sourced from Datadryad (https://doi.org/10.5061/dryad.h70rxwdmj) [32]. We employed SPATA software SPATA:: plotSurfaceInteractive and SPATA2 (https://github.com/theMILOlab/SPATA2, accessed on 20 March 2025) for the visualization of spatial gene expression. For spatial expression plots, either normalized and scaled gene expression values (to plot single genes) or enrichment scores of a defined gene set were used, with the 0.5 quantile of a probability distribution fitting. The x-axis and y-axis coordinates are given by the input file based on the localization to the H&E staining.
2.5. Cell Lines and Cell Culture
The following cell lines were utilized in this study: GSC models (MES28, GSC23, 387, 3691) generously donated by Dr. Jeremy N. Rich’s laboratory (University of Pittsburgh Medical Center, Pittsburgh, PA, USA), and neural stem cell lines (NSC11, ENSA, HNP1). All stem cell populations were maintained in serum-free DMEM/F12 medium (Gibco, Waltham, MA, USA) enriched with B27 supplement (Life Technologies, Carlsbad, CA, USA) and dual growth factors (20 ng/mL bFGF/EGF; R&D Systems, Minneapolis, MN, USA). Corresponding DGCs were propagated in DMEM basal medium (Gibco 11995065) containing 10% fetal bovine serum (Gibco 26140079) to preserve their differentiated phenotype.
2.6. SiRNA Transfection
ZEB1 siRNAs were purchased from Dharmacon (ON-TARGETplus Human ZEB1 siRNA, J-006564-10-0050, J-006564-11-0050). MMP9 siRNAs were purchased from Dharmacon (ON-TARGETplus Human MMP9 siRNA, J-005970-07-0050, J-005970-08-0050). ZO-1(TJP1) siRNAs were purchased from Dharmacon (ON-TARGETplus Human TJP1 siRNA, J-007746-05-0020, J-007746-06-0020). A total of 2,500,000 of GSC23 cells were electroporated with either control, ZEB1, MMP9, or ZO-1(TJP1) siRNA (100 nM) using nucleofector (kit C and program X-05). Then, the cells were plated in 5 mL of complete neurobasal media and incubated for 48 h at 37 °C until experiment analysis.
2.7. Immunoblotting
Protein extraction was performed with RIPA lysis buffer (P0013B, Beyotime, Shanghai, China) supplemented with protease inhibitors, and protein concentrations were determined using a BCA assay (Thermo Fisher Scientific, Waltham, MA, USA). Equal protein quantities from cellular or tissue lysates were separated on 10–17% SDS-PAGE gels. Following electrophoretic separation and membrane transfer, blots were probed with specific primary antibodies followed by Goat anti-Mouse or Goat anti-Rabbit IgG (H+L) HRP-linked secondary antibodies (31430 and 31460, Invitrogen, Waltham, MA, USA). Protein signals were detected by enhanced chemiluminescence (Clarity Western ECL Substrate, Bio-Rad, Hercules, CA, USA) and quantified through grayscale densitometry. A detailed list of primary antibodies is presented in Table 2. All the uncropped original blots are provided in Supplementary Figures S1 and S2.
2.8. Statistical Analysis
The statistical software R (version 4.0.3) and GraphPad Prism 10 software (version 10.1.0) were used for the statistical analysis and the generation of figures. The median values of EMT-related gene expression were considered the cutoff value to separate patients into the high and low groups. Survival analysis was conducted using Kaplan–Meier analysis and the log-rank test. The Wilcoxon signed rank test, Mann–Whitney test, and unpaired t-test were used for statistical analysis between two groups, while the Kruskal–Wallis test was applied for statistical analysis between more than two groups. The non-parametric Spearman test was conducted to evaluate the correlation of two variables. All statistical tests were independently performed by two different scientists, and a p-value of less than 0.05 was considered statistically significant.
3. Results
3.1. Initial Clustering and Identification of Cell Types in GBM
To investigate cell–cell communication patterns of GSCs in GBM, we analyzed a scRNA-seq dataset (GSE84465) from the GEO database containing 3589 cells derived from human primary GBM samples. We used k-means clustering on the 2D UMAP map, resulting in the identification of 10 distinct cell types within separate clusters, which includes GSCs, DGCs, oligodendrocyte precursor cells (OPCs), microglia, astrocytes, neurons, vascular cells, oligodendrocyte, monocytes/macrophages, and undefined cancer cells (Figure 1A).
3.2. Overview of the Communication Network in GBM
To infer and visualize intercellular communications in GBM, we ran CellChat analysis on the scRNA-seq dataset mentioned above. CellChat analysis revealed intricate cell–cell communication networks in GBM, with all pairwise combinations among the 10 cell clusters exhibiting varying degrees of interaction intensity, including notable autocrine signaling within individual cell populations (Figure 1B).
A total of 43 significant signaling pathways among the 10 cell groups were detected (Figure 1C). The signaling pathways associated with GSCs encompassed multiple molecular systems, including pleiotrophin (PTN), midkine (MK), secreted phosphoprotein 1 (SPP1), complement signaling, annexin-mediated pathways, visfatin-related mechanisms, fibroblast growth factor (FGF) signaling, colony-stimulating factor (CSF) pathways, prosaposin (PROS)-associated signaling, transforming growth factor beta (TGF-β) pathways, vascular endothelial growth factor (VEGF) signaling, leukemia inhibitory factor receptor (LIFR)-mediated communication, and calcitonin receptor (CALCR)-related interactions.
3.3. Cell–Cell Communication Strength Across 10 Cell Groups
Figure 2 illustrates the communication strength between each cell type and others, where GSCs demonstrate robust autocrine signaling (self-communication), followed by notable paracrine interactions with microglia, monocytes/macrophages, and astrocytes. Surprisingly, their communication intensities with neurons, oligodendrocytes, and DGCs were comparatively weaker.
3.4. The Cell Communication Pattern of GSCs Is Mostly Related to EMT
To further investigate how multiple cellular groups and signaling pathways coordinate their functions, we employed CellChat to identify global communication patterns using a pattern recognition approach. Through the selectK function, we inferred the optimal number of communication patterns to be six. The outgoing communication patterns revealed how sender cells (i.e., signal-originating cells) coordinate with each other and interact with specific signaling pathways to drive cellular communication. Among these six outgoing patterns, GSCs were predominantly associated with Pattern 1 (Figure 3A), which encompasses signaling pathways including MK, ANNEXIN, CSF, and LIFR (Figure 3B).
KEGG analysis of the genes associated with these pathways demonstrated significant enrichment in EMT processes (Figure 3C). Previous studies have demonstrated that MK promotes EMT-mediated cisplatin resistance [33]. Members of the ANNEXIN family, including ANXA2, can form complexes with TTK, which activate the Akt/mTOR signaling pathway and promote EMT [34]. G-CSF and CSF-1 were key factors, which promoted the EMT, migration, and invasion of trophoblasts via activating the PI3K/Akt/Erk1/2 signaling pathway [35,36]. LIFR, functioning as a target of KMT2D, epigenetically activates the PI3K/Akt pathway and drives EMT [37]. Subsequent GSEA of TCGA-GBM data further revealed that enhanced enrichment of EMT-related genes correlated with increased secretory protein production capacity (Figure 3D). These findings collectively suggest a strong association between secretory protein signaling in GSCs and EMT activation.
3.5. Spatial Co-Localization of EMT and GSCs in GBM
To investigate the spatial correlation between EMT expression patterns and GSCs, we analyzed spatial transcriptomic data from three GBM specimens [32] (Figure 4A–C). Intriguingly, our findings revealed striking spatial concordance among stem cell proliferation, protein secretion activity, and EMT activation patterns.
3.6. Elevated Expression of EMT-Associated Genes in GBM
Subsequently, we sought to investigate the expression patterns of EMT-related genes in GBM. Given the extensive number of EMT-associated genes, we selected eight representative genes (MMP2, MMP9, SNAI2, VIM, CTNNB1, BIRC5, POSTN, CXCR4) for analysis [38]. Their expression profiles were analyzed using Gliovis platform within The Cancer Genome Atlas (TCGA) database. Comparative analysis revealed significant upregulation of all eight examined genes in GBM tissues compared to non-tumor controls (Figure 5). The majority of EMT-related genes in Pattern 1 of Figure 3C were also highly expressed in TCGA-GBM (Supplementary Figure S3).
3.7. Elevated Expression of EMT-Associated Genes Correlates with Poor Prognosis
Next, we investigated the prognostic relevance of these highly expressed genes in glioma patients using the Chinese Glioma Genome Atlas (CGGA) database. Our analysis revealed that all eight genes exhibited significantly shorter overall survival times in high-expression groups compared to their low-expression counterparts, suggesting a negative correlation between these genes and patient prognosis (Figure 6).
3.8. GSCs Exhibit Pronounced Overexpression of EMT-Associated Gene Signature in GBM
To compare the expression of EMT-related genes between GSCs and other GBM cells, we analyzed an additional scRNA-seq dataset comprising 28 early-passage GSC cultures derived from 24 patients and 14,207 malignant cells from seven patients with GBM [39] (Figure 7A). Our analysis revealed significantly higher expression levels of EMT-associated genes in GSCs compared to other GBM cell populations (Figure 7B).
The protein expression levels of BIRC5, VIM, CTNNB1, and SNAI2 were demonstrated by immunoblot analysis to be significantly elevated in four GSC lines (MES28, GSC23, 387, 3691) compared to their DGC counterparts and three NSC lines (NSC11, ENSA, HNP1) (Figure 7C). In two pairs of matched GSCs and DGCs analyzed by H3K27ac ChIP-seq, the H3K27ac peaks of BIRC5, VIM, CTNNB1, and SNAI2 were consistently elevated in GSCs compared to their corresponding DGC counterparts (Figure 7D–G). This observation suggests that EMT-associated genes exhibit enhanced expression in GBM, particularly within the GSC population.
3.9. EMT-Related Genes Are More Enriched in MES-like GSCs
As previously demonstrated, a subset of EMT-related genes exhibits elevated expression in GSCs, and the EMT signatures demonstrate a positive correlation with the GSC signatures (Figure 8A).
It was reported that different states of GSCs that are thought to act as key cancer drivers can spread tumors and resist radiation/chemo treatments better than other cells [8,40]. Cyril Neftel and colleagues categorized GSCs into four distinct phenotypic states based on cellular markers [14]: OPC-like, NPC-like, AC-like, and MES-like, which demonstrates the diversity of proliferating cells. To investigate the association between EMT-related genes and different GSC states, we performed ssGSEA analysis on those four GSC subtypes (GSE131928). The results revealed that MES-like GSCs exhibited the highest enrichment scores for EMT-related genes (Figure 8B,C), indicating their strongest correlation with the mesenchymal state. In the TCGA-GBM database, EMT-related genes also demonstrated significant correlations with MES-like GSC signatures (Figure 8D).
To confirm that EMT activation precedes MES differentiation in GBM progression, we transfected GSC23 cells with siRNAs targeting the most common EMT markers, e.g., ZEB1, ZO-1, and MMP-9, and subsequently examined the protein levels of CD44, a marker of the mesenchymal GSC subtype (Figure 8E–G). The results demonstrated that knockdown of EMT-related genes significantly reduced CD44 levels, further confirming that EMT activation precedes mesenchymal differentiation.
3.10. Clinical Significance: Worse Prognosis Associated with High MES-like Signature Expression in Brain Tumor Patients
Given the close association between EMT-related genes and MES-like GSCs, we next sought to investigate the clinical significance of MES-like signatures. Studies of intertumoral heterogeneity based on bulk expression profiles suggest that three subtypes of GBM exist, namely proneural (PN), classical (CL), and mesenchymal (MES) [41,42]. We performed survival analyses on all brain tumor patients [43] using the Brain TIME platform with these three subtypes of signatures (Figure 9A–C) and found that only the MES-like group demonstrated a significantly worse prognosis when exhibiting high expression levels (p = 0.0371).
4. Discussion
This study explored the communication pattern of GSCs in GBM. Existing studies have investigated cell–cell communication in GBM [44,45,46,47,48]; however, the majority have predominantly focused on elucidating the interactions between GSCs and immune cell populations [48,49]. For example, Cao et al. found that the MIF pathway was active between GSCs and myeloid-derived suppressor cells (MDSCs) [48]. GSCs may activate MDSCs to inhibit immune responses by secreting MIF. Li et al. identified important ligand–receptor interactions, including MIF binding with CD74 + CXCR4 or CD74 + CD44 and SPP1 binding with CD44 or ITGAV + ITGB1 [49]. They also highlighted the key role and higher interaction between monocytes/macrophages and MES-like malignant cells in the SPP1 signaling pathway, which is consistent with our results (Figure 1C). In our study, we focused on the most relevant biological functions of GSC signal pathways after enrichment analysis. Through KEGG analysis, we found that the cell communication patterns of GSC are most closely associated with EMT, suggesting that GSCs may play a role in promoting the mesenchymal transition of GBM (Figure 3).
The process of EMT involves a series of biochemical transformations in which epithelial cells, originally organized in a polarized sheet, acquire mesenchymal characteristics. This transition results in cells that exhibit reduced intercellular adhesion, increased mobility, and a mesenchymal phenotype. Additionally, these transformed cells develop distinct morphological features, enhanced resistance to apoptosis induced by detachment (anoikis), and increased tolerance to chemotherapy [50,51].
Because GBMs are not typical epithelial cells due to the absence of a basal membrane and inconsistent expression of E-cadherin, to coin the term “EMT” within the GBM context would be scientifically inaccurate. However, studies in recent years have found that the regulation of the classical EMT markers can induce the GBM cells to the invasive MES subtype [52,53]. Compared to bulk RNA-seq approaches in prior studies by Neftel et al. [14], we used CellChat to infer cell–cell communication and revealed that the expression of EMT-associated genes may serve as a driving force for MES-like phenotypic transformation, suggesting that these genes may act as transcriptional regulators of the MES phenotype. Our findings also indicate that EMT-related genes are more enriched in MES-like GSCs (Figure 8). These findings highlight the parallels in genetic profiles shared by both the MES transition and the classical EMT process, encompassing key elements such as traditional EMT markers, transcription factors, and the activation of signaling pathways.
Our study also revealed that MES signatures are associated with poorer prognostic outcomes in brain tumor patients compared with PN or CL signatures (Figure 9A). The PN and MES expression subtypes have been most consistently described in the literature, with PN relating to a more favorable outcome and MES relating to poor survival [54,55,56], which is consistent with our results with different datasets (Figure 9A,B). MES-like glioma cell differentiation status has been found to correlate with enrichment of macrophages/microglia [41,57,58]. It is also reported that EMT is the main way in which glioma-associated microglia/macrophages (GAMs) promote glioma progression [59]. These findings suggest that GSCs may facilitate MES differentiation through EMT-associated cell communication patterns, subsequently recruiting immunosuppressive GAMs. This cascade could contribute to GBM immune evasion, thereby promoting tumor progression. However, further experimental validation is required to substantiate these conclusions.
This study also has certain limitations and shortcomings. Firstly, the scRNA-seq database we utilized for CellChat contained a relatively small number of cells (n = 3589). To further validate our conclusions, it will be necessary to employ larger databases with greater cell numbers in future studies. Second, we relied on published retrospective datasets, necessitating future validation in a prospective, large cohort. Third, we should conduct more in-depth and detailed molecular biology studies in both in vivo and in vitro experiments to uncover the molecular mechanisms of MES differentiation of GSCs and identify new therapeutic targets.
5. Conclusions
Our study suggests that GSCs facilitate GBM progression through intercellular communication in the pattern of EMT. EMT-associated genes may drive the differentiation of GSCs toward a MES-like phenotype, thereby leading to poorer clinical outcomes. Consequently, targeting EMT-related pathways could represent a novel therapeutic strategy for GBM treatment.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ostrom Q.T. Price M. Neff C. Cioffi G. Waite K.A. Kruchko C. Barnholtz-Sloan J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019 Neuro-Oncology 202224 v 1v 9510.1093/neuonc/noac 20236196752 PMC 9533228 · doi ↗ · pubmed ↗
- 2Delgado-López P.D. Corrales-García E.M. Survival in Glioblastoma: A Review on the Impact of Treatment Modalities Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex.2016181062107110.1007/s 12094-016-1497-x 26960561 · doi ↗ · pubmed ↗
- 3Alexopoulos G. Zhang J. Karampelas I. Patel M. Kemp J. Coppens J. Mattei T.A. Mercier P. Long-Term Time Series Forecasting and Updates on Survival Analysis of Glioblastoma Multiforme: A 1975-2018 Population-Based Study Neuroepidemiology 202256758910.1159/00052261135172317 · doi ↗ · pubmed ↗
- 4Omuro A. De Angelis L.M. Glioblastoma and Other Malignant Gliomas: A Clinical Review JAMA 20133101842185010.1001/jama.2013.28031924193082 · doi ↗ · pubmed ↗
- 5Stupp R. Mason W.P. van den Bent M.J. Weller M. Fisher B. Taphoorn M.J.B. Belanger K. Brandes A.A. Marosi C. Bogdahn U. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma N. Engl. J. Med.200535298799610.1056/NEJ Moa 04333015758009 · doi ↗ · pubmed ↗
- 6Prager B.C. Bhargava S. Mahadev V. Hubert C.G. Rich J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos Trends Cancer 2020622323510.1016/j.trecan.2020.01.00932101725 PMC 8779821 · doi ↗ · pubmed ↗
- 7Zhou B.-B.S. Zhang H. Damelin M. Geles K.G. Grindley J.C. Dirks P.B. Tumour-Initiating Cells: Challenges and Opportunities for Anticancer Drug Discovery Nat. Rev. Drug Discov.2009880682310.1038/nrd 213719794444 · doi ↗ · pubmed ↗
- 8Bao S. Wu Q. Mc Lendon R.E. Hao Y. Shi Q. Hjelmeland A.B. Dewhirst M.W. Bigner D.D. Rich J.N. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response Nature 200644475676010.1038/nature 0523617051156 · doi ↗ · pubmed ↗