Application of a high-resolution melt assay for monitoring SARS-CoV-2 variants in Burkina Faso and Kenya
Caitlin Greenland-Bews, Sonal Shah, Morine Achieng, Emilie S. Badoum, Yaya Bah, Hellen C. Barsosio, Helena Brazal-Monzó, Jennifer Canizales, Anna Drabko, Alice J. Fraser, Luke Hannan, Sheikh Jarju, Jean-Moise Kaboré, Mariama A. Kujabi, Cristina Leggio, Maia Lesosky, Jarra Manneh

TL;DR
This study introduces a low-cost, high-throughput method to monitor SARS-CoV-2 variants in low-resource settings using HRM assays, validated in Burkina Faso and Kenya.
Contribution
The development of two HRM assays for variant detection in low-resource settings, validated against sequencing data.
Findings
HRM-VOC-1 and HRM-VOC-2 assays showed high sensitivity and specificity for detecting Alpha, Delta, and Omicron variants.
The HRM-VOC-2 assay was scaled to screen 506 samples, revealing the replacement of Alpha by Delta and then by Omicron in Kisumu, Kenya.
The HRM assays provide a cost-effective alternative for variant surveillance in areas with limited sequencing infrastructure.
Abstract
The rapid emergence and global dissemination of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) highlighted a need for robust, adaptable surveillance systems. However, financial and infrastructure requirements for whole-genome sequencing mean most surveillance data have come from higher-resource geographies, despite unprecedented investment in sequencing in low- and middle-income countries (LMICs). Consequently, the molecular epidemiology of SARS-CoV-2 in some LMICs is limited, and there is a need for more cost-accessible technologies to help close data gaps for surveillance of SARS-CoV-2 variants. To address this, we have developed two high-resolution melt (HRM) curve assays that target variant-defining mutations in the SARS-CoV-2 genome, which give unique signature profiles that define different SARS-CoV-2 variants of concern (VOCs). Extracted RNA from SARS-CoV-2-positive…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
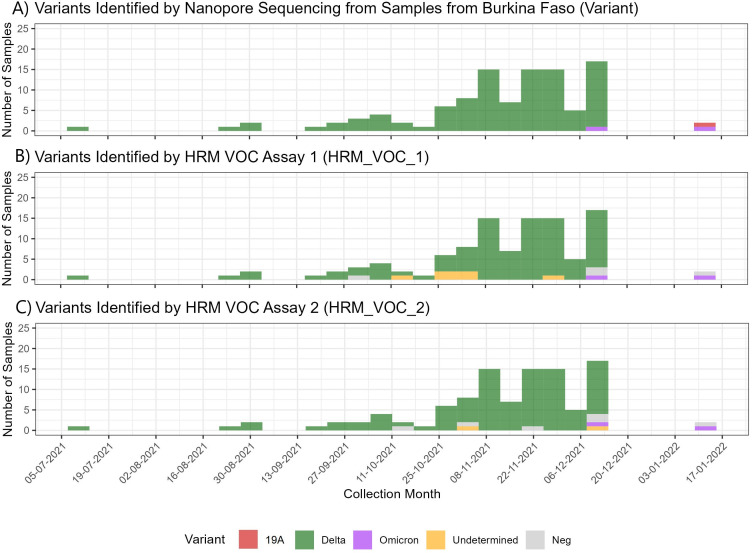 Fig 1
Fig 1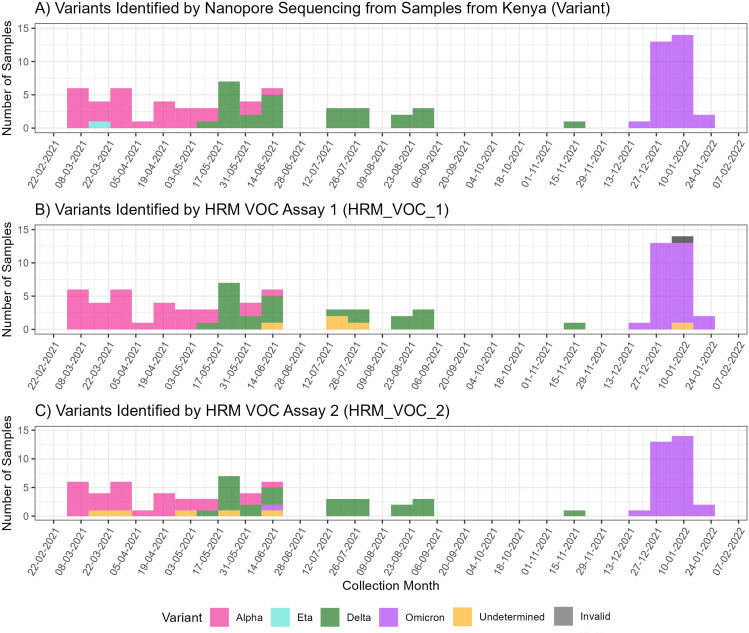 Fig 2
Fig 2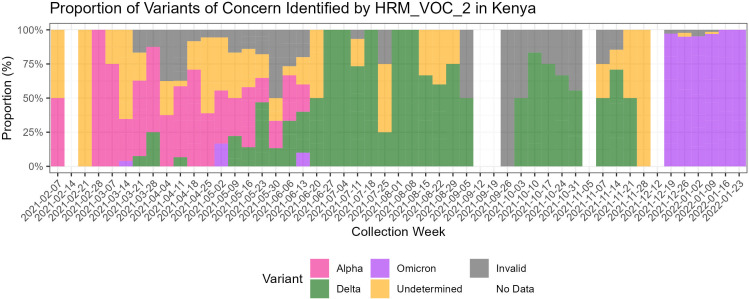 Fig 3
Fig 3| Assay | Mutation targeted | Associated variants | Final forward primer concentration (nM) | Final reverse primer concentration (nM) |
|---|---|---|---|---|
| HRM-VOC-1 | S_del. 156–157 | Delta | 100 | 100 |
| S_K417N | Beta/Gamma/Omicron BA.1 | 150 | 150 | |
| N_D3L | Alpha | 600 | 600 | |
| S_EPE | Omicron BA.1 | 250 | 250 | |
| HRM-VOC-2 | S_A570D | Alpha | 400 | 400 |
| S_L452R | Delta | 200 | 200 | |
| S_EPE | Omicron BA.1 | 400 | 400 | |
| Orf1b_Control | All | 100 | 100 |
| Assay and VOC | True positive | True negative | False positive | False negative | Sensitivity (%) [CI: 95%] | Specificity (%) [CI: 95%] | Accuracy (%) |
|---|---|---|---|---|---|---|---|
| HRM-VOC-1 | |||||||
| Alpha | 28 | 164 | 1 | 0 | 100 [87.66–100] | 99.39 [96.67–99.98] | 99.48 |
| Delta | 118 | 62 | 0 | 13 | 90.08 [83.63–94.61] | 100 [94.22–100] | 93.26 |
| Omicron | 30 | 161 | 0 | 2 | 93.75 [79.19–99.23] | 100 [97.73–100] | 98.96 |
| HRM-VOC-2 | |||||||
| Alpha | 26 | 164 | 0 | 2 | 92.86 [76.50–99.12] | 99.39 [97.78–100] | 98.96 |
| Delta | 120 | 62 | 0 | 10 | 92.31 [86.31–96.25] | 100 [94.22–100] | 94.79 |
| Omicron | 32 | 159 | 1 | 0 | 100 [89.11–100] | 99.38 [96.57–99.98] | 99.48 |
| Variant | McNemar’s chi-squared | df | |
|---|---|---|---|
| Alpha | 1.33 | 1 | 0.25 |
| Delta | 0.08 | 1 | 0.77 |
| Omicron | 0.5 | 1 | 0.48 |
| Assay and variant | Cohen’s kappa |
|---|---|
| HRM-VOC-1 | |
| Alpha | 0.81 |
| Delta | 0.68 |
| Omicron | 0.79 |
| HRM-VOC-2 | |
| Alpha | 0.81 |
| Delta | 0.70 |
| Omicron | 0.79 |
- —UKPHRST
- —National Environment Research Council
- —U.S Food and Drug Administration Medical Countermeasures Initiative Contract
- —National Institute for Health Research Health Protection Research Unithttp://dx.doi.org/10.13039/100018336
- —Bill and Melinda Gates Foundationhttp://dx.doi.org/10.13039/100000865
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSARS-CoV-2 and COVID-19 Research · SARS-CoV-2 detection and testing · vaccines and immunoinformatics approaches
INTRODUCTION
As the coronavirus disease 2019 (COVID-19) pandemic progressed, the evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) gave rise to variants of concern (VOCs). These VOCs posed an increased and significant threat to the global population and jeopardized public health measures and interventions that had been deployed (1). Detection and surveillance of these variants were primarily achieved through sequencing, which was crucial for tracking the spread of the VOCs worldwide. Genomic surveillance is only beneficial when it is representative spatially and temporally (2), and while many countries benefitted from real-time genomic surveillance during the COVID-19 pandemic, most genomic information of SARS-CoV-2 is from higher-income countries (3).
As of September 2021, 18 months into the COVID-19 pandemic, sequences originating from Africa accounted for approximately 1% of the total 3.5 million sequences available (4). Similarly, it was found that as of October 2021, high-income countries were uploading 12 times more sequences than low- and middle-income countries (2). As of March 2022, there were 100,000 SARS-CoV-2 sequences available from African countries. This represented an incredible milestone in genomic surveillance in Africa and is the result of huge investments to increase sequencing capacity, with SARS-CoV-2 sequences far outnumbering any number of pathogen sequences submitted before from the continent (2). Although investments in sequencing infrastructure are ongoing, this surveillance gap highlights the need for more accessible surveillance methods to be developed and utilized in the interim. Molecular diagnostics offer a viable alternative for targeting SARS-CoV-2 VOCs that are highly sensitive.
One promising method is high-resolution melt (HRM) assays, which feature a post-PCR analysis method that is highly sensitive in detecting nucleotide changes from shifts in amplicon melting temperature. This method has been used to identify individual mutations (5–9) with high sensitivity for detecting their respective targets. The broad range of mutations targeted across the literature includes the VOC-specific mutations N501Y, D614G, L452R, and K417N/T (7–9). However, many of these assays must be run simultaneously in singleplex to allow differentiation between multiple VOCs. This increases the work time, cost of reagents, and the volume of valuable samples required for genotyping. A one-step HRM that could identify multiple mutations in one assay while cutting down on cost and time would be ideal.
Here, we build upon our previous work of one such HRM assay capable of identifying Alpha, Beta, Gamma, Delta, and Omicron VOCs (10). We have developed our toolkit approach further, expanding the available primer sets and developing a new assay that targets the Alpha, Delta, and Omicron (BA.1) VOCs. We evaluate both assays’ ability to detect Alpha, Delta, and Omicron (BA.1) VOCs and compare our HRM results against next-generation sequencing (NGS) from Oxford Nanopore MinION (MK1B, Oxford, UK). This evaluation was conducted using samples collected in Burkina Faso and Kenya from February 2021 to February 2022.
MATERIALS AND METHODS
Sample collection and study setting
All samples were collected as part of the Malaria as a Risk Factor for COVID-19 in Western Kenya and Burkina Faso (MALCOV) study (NCT04695197). Mid-nasal swabs were taken from SARS-CoV-2-positive participants and stored in viral transport media (Biocomma). Samples were collected between February 2021 and February 2022. Details of the study settings and sites involved can be found in the study protocol (11). This study was conducted across locations in the United Kingdom and sub-Saharan Africa. Assays were developed and validated in the United Kingdom, training was conducted in Kenya and Gambia, and testing was conducted in Kenya, Gambia, Burkina Faso, and the United Kingdom. One hundred twelve samples from Burkina Faso and 93 from Kenya were sequenced and analyzed by both HRM assays (HRM-VOC-1 and HRM-VOC-2). A further 413 samples from the Kenyan cohort were analyzed by HRM-VOC-2 (total sample count analyzed by HRM-VOC-2, n = 506) but were not sequenced to determine the molecular epidemiology of the variants of concern.
RNA extraction
RNA was extracted from clinical specimens in viral transport media (VTM) using the QIAamp Viral RNA Kit (QIAGEN, Germany), following the manufacturer’s protocol, and implemented as an automated workflow using the QIAcube HT platform (QIAGEN, Germany). Purified RNA was eluted in 50 µL of elution buffer and stored at −80°C until use.
Reverse transcription-polymerase chain reaction (RT-PCR)
RT-PCR was performed by staff on-site in Kenya and Burkina Faso according to the study protocol.
Design of HRM-VOC-2 assay
Sequences representing the known variants classified by the World Health Organization as variants of concern (VOC), under monitoring (VUM), and of interest (VOI) were downloaded from GenBank and aligned using ClustalX in BioEdit (version 7.2.5). Lineage-defining mutations were identified from the literature and online repository https://covariants.org/ (12) and located within the alignment.
Primers were designed (Table S1) with the aid of Primer 3 (13), and where no suitable primers could be obtained, primers were designed manually. The suitability of primers was initially tested in silico using OligoCalc (14) and uMelt (15) to ensure compatible melting temperatures (Tms).
Singleplex testing was conducted during assay development to ensure specificity of primers and was conducted by testing each primer pair on extracted RNA from cultured viral isolates for Alpha (GenBank accession number: MW980115), Beta (hCoV-19/South Africa/KRISP-EC-K005321/2020) (BEI Resources), Gamma (hCoV-19/Japan/TY7-503/2021), Delta (SARS-CoV-2/human/GBR/Liv_273/2021), Omicron (BA.1) (SARS-CoV-2/human/GBR/Liv_1326/2021), and wild type (isolate REMRQ0001/Human/2020/Liverpool) (Alpha/Beta/Gamma/Delta/OmicronBA.1/OmicronBA.2/WT), and following this, a multiplex was formed with compatible peak Tms that targeted Alpha, Delta, and Omicron (BA.1). No further testing was possible with Beta and Gamma variants due to their absence in the clinical sample sets.
HRM assays
Two multiplex HRM assays were evaluated, each containing four different primer pairs, the HRM-VOC-1 assay as described in (10) and the HRM-VOC-2 assay described above. For each assay, 2.5 µL of RNA template was added for 12.5 µL final reaction volumes using Lunar Universal Probe One-Step RT-qPCR kit (New England BioLabs, USA), 1× EvaGreen dye (Biotium, USA), and primers added to their optimized concentrations (Table 1).
Reactions were performed using QuantStudio 5 (Thermo Fisher, USA) for Kenyan samples and QuantStudio 6/7 flex (Thermo Fisher, USA) for Burkinabe samples. The thermal cycle profiles are found in Table S2.
Analysis of HRM assay data
Data were visualized as negative first derivative plots using QuantStudio Design and Analysis Software (v.1.5.2, QuantStudio 5 systems, Thermo Fisher Scientific Inc.).
Samples that did not yield enough sequence coverage by nanopore sequencing to identify a variant using NextClade (16) were excluded from further analysis. Samples that gave an HRM peak that could not be assigned to a variant were classified as undetermined. In the instance of HRM-VOC-2, where there is a control peak, if the control peak was absent, these samples were classified as invalid. For HRM-VOC-2, if there is a control peak but the remaining peaks do not fit the signature peaks for the variants of concern and therefore cannot be assigned, these samples were classified as undetermined. For analysis of the assay performance, invalid HRM results were excluded ([Fig. 1 and 2](#F1 F2)). Sensitivity and specificity analysis was performed in the MedCalc diagnostics calculator (17).
SARS-CoV-2 variants identified in Burkina Faso using different methods. (A) Number of samples collected in Burkina Faso from July 2021 to January 2022 and the variants that were identified by nanopore sequencing. (B) Number of samples in the Burkina Faso cohort and the variant identified by using the HRM-VOC-1 assay. Negative results represent those with no amplification observed; undetermined samples had amplification but no identifiable VOC peak. (C) Number of samples in the Burkina Faso cohort and the variant identified by using the HRM-VOC-2. Negative results are those where no amplification was observed, and undetermined results are those with a control peak without an identifiable VOC peak.
SARS-CoV-2 variants identified in Kenya using different methods. (A) Number of samples collected in Kenya from February 2021 to February 2022 and the variants that were identified by nanopore sequencing. (B) Number of samples in the Kenyan cohort and the variant identified by using the HRM-VOC-1 assay. Negative results represent those with no amplification observed; undetermined samples had amplification but no identifiable VOC peak. (C) Number of samples in the Kenyan cohort and the variant identified using the HRM-VOC-2. Negative results represent those with no amplification observed; undetermined samples had amplification but no identifiable VOC peak.
Sequencing
Two hundred five SARS-CoV-2 samples from Burkina Faso and Kenya combined were prepared according to the Artic SARS-CoV-2 sequencing protocol (18). Amplicon generation was conducted using Artic vV.4.1 primers (Integrated DNA Technologies, USA), using Q5 Hot Start High-Fidelity 2× Master Mix (New England Biolabs, USA), 10 µM primer pools, and a thermocycling profile of 30 seconds of 98°C heat inactivation, followed by 25 cycles of 15 seconds of denaturation at 98°C and 5 minutes of annealing/extension at 65°C. Library preparation was carried out using the Ligation Sequencing Kit (SQK- LSK109) and Native Barcoding Expansion Kits (EXP-NBD196, Oxford Nanopore Technologies, UK). Enzymes for barcode and adapter ligation were acquired from New England Biolabs (USA), and AMPure XP beads were acquired from Fisher Scientific (USA). Sequencing was performed on an R.9.4.1 flow cell on a MinION Mk1B device (Oxford Nanopore Technologies, UK) for Kenyan samples and GridION device for Burkinabe samples. All sequences have been deposited on the Sequence Read Archive under the BioProject numbers PRJNA1095865 and PRJNA1096688 for sequences from Kenya and Burkina Faso, respectively.
Sequencing analysis/bioinformatics
Bioinformatics analysis was performed by following the Artic bioinformatics pipeline (v.1.1.0) (19). Basecalling was performed using Guppy, and a consensus sequence was generated. Consensus sequences were processed by NextClade (v.2.14.1) (16) for rapid variant calling and mutation summaries.
Statistical analysis and data processing
Diagnostic accuracy
Samples that did not yield enough coverage from sequencing for a variant to be identified were excluded from analysis due to the lack of a reference standard.
Sensitivity, specificity, and accuracy were calculated for each variant by comparison to the reference standard NGS, and the calculation was performed using MedCalc (17). True positives were defined as samples where the HRM-identified variant matched the variant identified by sequencing. A true negative was every sample correctly identified as a variant other than the target VOC for that analysis. Overall agreement with the NGS result was calculated per assay as the total number of true positives divided by the total number of samples sequenced. Cohen’s kappa (agreement) was calculated and interpreted per variant for each assay as described in McHugh et al. (20).
Comparison of HRM-VOC-1 and HRM-VOC-2
McNemar’s test was applied to compare the results of HRM-VOC-1 and HRM-VOC-2. This test was performed for each variant (Alpha, Delta, Omicron) using the mcnemar.test() function in R.
Data processing and visualization
All data visualization was conducted using R in RStudio (version: 2023.3.1.446). Graphical analysis was undertaken using the ggplot2 package.
RESULTS
Variants identified in Kisumu, Kenya
Samples were collected in Kisumu, Kenya, from February 2021 to February 2022 (Fig. 2). In the sample set of 93 samples, nanopore sequencing identified six different clades, 20I (n = 28), 21D (n = 1), 21A (n = 20), 21J (n = 5), 21I (n = 2), 21K (n = 30), corresponding to four different variants, Alpha, Eta, Delta, and Omicron, and six Pango lineages (Fig. S2). Seven samples did not yield high enough quality reads for a variant to be identified using NextClade.
Detection by HRM
Of the 193 samples with a valid sequencing result from both settings combined, the HRM-VOC-1 assay identified variants in 176 of these (91.2%). One hundred eighteen were identified as Delta, 30 as Omicron, and 28 as Alpha. Eleven samples produced a peak profile, but a variant could not be determined, and five samples showed no amplification and were classified as negative (Fig. 2). The HRM-VOC-2 assay identified variants in 179 of 193 samples (92.7%), 120 were Delta variants, 33 were Omicron, and 26 were Alpha. There was one sample identified as Eta by sequencing; this was identified as a false-positive Alpha result by HRM-VOC-1 and gave a peak classified as “unidentified” by HRM-VOC-2. Seven samples gave a positive HRM result but did not have a peak profile indicative of one of the three targeted VOCs (Alpha/Delta/Omicron), six showed no amplification and were classed as negative, and there was one invalid sample result (Fig. 2). Invalid results were not included in graph visualization or sensitivity or specificity analysis.
Assay performance
Sensitivity and specificity were calculated for the combined HRM results across both study locations in comparison with NGS reference (Table 2). One hundred ninety-three samples gave a valid result when using the HRM-VOC-1 assay, and 192 samples when using the HRM-VOC-2 assay. The HRM-VOC-1 assay had a sensitivity and specificity of 100% and 99.39%, respectively, for Alpha, 90.08% and 100% for Delta, and 93.75% for Omicron. The HRM-VOC-2 assay had a sensitivity and specificity of 92.86% and 99.39%, respectively, for Alpha, 92.31% and 100% for Delta, and 100% and 99.38% for Omicron.
McNemar’s and Cohen's kappa test results
No significant difference was found between the two HRM assays for detecting the three key variants using McNemar’s test (Table 3).
There was substantial agreement with the sequencing results for both HRM-VOC-1 and HRM-VOC-2, detecting Delta and Omicron, as Cohen’s kappa was between 0.61 and 0.80, and there was almost perfect agreement with sequencing for Alpha samples (Table 4)(20).
Cycle threshold (Ct) value vs sequencing and HRM success
All samples analyzed were below RT-qPCR Ct 30, with the majority being successfully called by both assays (Fig. S3). One sample gave an invalid result for HRM-VOC-1 with a Ct of 24. Nine samples gave an undetermined result for HRM-VOC-1 with a Ct range of 23.5–29.5, and 10 were undetermined by HRM-VOC-2 and had a Ct range of 23.5–29.5 in both instances.
Scaling up sample screening by HRM in Kenya
Out of the 506 positive SARS-CoV-2 samples analyzed by HRM-VOC-2, 396 had an identifiable variant (78.3%) (Fig. 3). Of the identifiable variants, 72 samples (18.18%) were identified as Alpha, 98 samples (24.75%) were identified as Delta, and 226 samples (57.07%) were identified as Omicron. Of the remaining 110 samples, 47 gave invalid peak readings (no or limited amplification observed and absence of a control peak), 63 amplified with a control peak, but the other peaks could not be categorized into signature peaks representing the variants of concern and therefore have been labeled as “undetermined” (Fig. 3). Cts were obtained from the MALCOV study team, and it was determined that of these 506 samples, Cts ranged from 17.9 to 39.9, with variants being successfully called across this range (Fig. S4). Samples that could not be called and were labeled as invalid (Fig. 3) all had a Ct of 30 or above, and undetermined samples had a range of 23.5–39.2 (Fig. S4).
Time series of variants identified by HRM-VOC-2 when tested on 506 SARS-CoV-2 PCR-positive samples collected in Kenya throughout the study period. This is a combined data set including the 86 successfully sequenced samples. Undetermined represents samples that produced a control peak but no identifiable VOC peaks. Invalid represents samples where there was no control peak. No data represents weeks of the year where no positive samples had been collected.
Alpha was the dominant variant in the data set at the start of sample collection (7 February 2021) until early May (2 May 2021). From the 2nd of May 2021, the proportion of detected Delta samples increased rapidly. As of 27 June 2021, Delta samples comprised 100% of samples collected. Delta remained the dominant variant detected until the 19th of December when Omicron fully replaced Delta at just below 100% of the total samples analyzed. Three samples were identified as Omicron by HRM-VOC-2 in the first 6 months of the sample set and are represented as Omicron samples in Fig. 3. Due to the timings of these samples being collected, implying it is unlikely that Omicron was circulating at this time, the decision was made to label these three samples as false positives.
DISCUSSION
Here, we have presented the application of two variant-calling HRM assays to the genotyping of positive SARS-CoV-2 samples in Burkina Faso and Kenya. These assays had high sensitivity in identifying the Alpha, Delta, and Omicron (BA.1) variants of concern when compared to results generated by NGS as the gold standard. Most samples were successfully variant-typed with high concordance to NGS results.
The assay was successfully scaled up to screen more than 500 samples collected over 12 months for the MALCOV study in Kisumu, Kenya. With the HRM assay, we could identify the infecting variant in many of these samples and describe the variant waves in Kisumu during this time. The ability to successfully scale up the HRM demonstrates that if implemented, the HRM can be used for high-throughput analysis of samples that could not be achieved with sequencing. When experiencing high case numbers, this scalability would aid in reducing backlogs and allow prioritization of any existing sequencing infrastructure to be used for samples that were unable to be identified by HRM. Furthermore, the speed of the HRM workflow and analysis is considerably faster than sequencing (a few hours compared to a few days, respectively), which would improve the time to generate crucial data to be used for public health data but would also free the time of laboratory staff for other important tasks.
Our assay has shown three variant replacement events between February 2021 and January 2022, which mirrors the three waves reported during this period from other African countries such as The Gambia (21), Ethiopia (22), and Senegal (23). In the samples analyzed from Kenya in this study, Alpha was the dominant variant between March 2021 and May 2021 and was then replaced by Delta in May 2021, followed by Omicron in mid-December 2021, which is in keeping with epidemiological data from other regions of Kenya (24, 25). Other studies have reported the Beta variant co-circulating with the Alpha variant in regions of Kenya (26). The HRM-VOC-2 assay used to screen all 506 samples in this study does not detect the Beta VOC, and no samples were identified as Beta in those that were sequenced, so it is unknown whether Beta was present in this sample set.
We have demonstrated that HRM is a reliable method of generating epidemiologically important data. HRM assays are also easily scalable, with 506 samples being variant-typed by the HRM-VOC-2 assay. Samples with a Ct lower than or equal to 30 provide the best results for identifying a VOC when analyzing with HRM. When testing the 506 samples, all invalid results for HRM had a Ct greater than 30 (Fig. S4), indicating that samples with lower Ct values should be prioritized where possible to minimize invalid results. Invalid and unidentifiable results when using the HRM are to be expected, even with samples with a Ct lower than 30; however, we recommend that the samples that are either unidentifiable variants or give invalid results with HRM could then be prioritized for NGS. Due to the volume of samples, it would have been expensive and labor-intensive to sequence the total sample set. Based on our results, this would result in only 20% of the total sample set requiring sequencing, reducing the overall expenditure. Finally, the calculated cost of this assay equates to <12 per sample when performed at high throughput (19). If scaling testing up to 500 samples using the HRM would cost ~6,000 for NGS, this would equate to ~$5,500 savings if using HRM.
Several HRM panels for variant identification have been developed throughout the pandemic. An HRM panel for detecting Delta, Omicron BA.1, BA.2, and BA.5 in four separate HRM assays achieved 97.9% agreement with Sanger sequencing (27). Another study in Iran utilized HRM for variant typing due to limited funds available for extensive sequencing and saw 93.68% sensitivity and 100% specificity compared with Sanger sequencing (6). The advantage of the approach presented here is the use of a single-tube assay that can detect Alpha, Delta, and Omicron variants in one reaction without requiring multiple tests, reducing test complexity.
Throughout this study, the assays have been run on multiple instruments when used at different study sites, including QuantStudio 5, QuantStudio 6/7 (Thermo Fisher Scientific Inc, USA), Magnetic Induction Cycler (MIC) (BioMolecular Systems, Australia), and Rotorgene Q (QIAGEN, Germany), highlighting the adaptability of the assays to multiple platforms. The transferability of this assay across platforms negates the need for instrument procurement if deciding to implement this technique, as most modern thermocyclers with the capability to perform HRM can be used.
HRM identified a small number of samples as Omicron in the first 6 months of the sample set. As this is a retrospective sample set, we identified these as probable false-positive results as they pre-date the established date of the first global report of Omicron BA.1 and the date of first detection in Kenya, both occurring in early November 2021 (26, 28). This could be due to non-specific binding of the Omicron primer sets to the RNA, or alternatively, a mutation in one of the primer target sites resulting in a temperature shift of the peak that results in the shift of a peak into the Tm range for Omicron for HRM-VOC-2 resulting in the miscalling of the Omicron variant for these samples. To fully understand these false positives, sequencing would need to be performed to investigate the potential mutations present in the target regions; however, this was outside the scope of this study at the time it was conducted.
The main limitation of this approach, which has also been noted across the literature, is its inability to detect new, emerging mutations, as the assay design relies on pre-existing knowledge of the mutation profile of circulating variants. However, from existing whole-genome sequencing (WGS) surveillance systems, information on novel single nucelotide polymorphisms (SNPs) of novel VOCs can be utilized in the design of HRMs to provide a more agile and accessible assay for more regions to have ownership of their surveillance efforts. In addition to this, unusual peaks from the HRM assays may be observed as a result of new, emerging mutations, and these unusual results can act as a flag for samples to be investigated further by sequencing, allowing the prioritization of samples and avoiding overburdening of existing sequencing infrastructures.
Another limitation lies in the inter-assay variation, which can impact assay interpretation when the assay relies on small shifts in melting temperatures. To improve the analysis of HRM outputs, automation of the process could be used to reduce any user error/unreliability in peak interpretation, which could be achieved through machine learning methods that use previously analyzed data sets to train an algorithm to interpret future outputs (29). This technology could be adapted to provide molecular epidemiological information on other pathogens without the expense of WGS.
While interest in surveillance of SARS-CoV-2 VOCs is waning (as of writing in 2025), this study has demonstrated the power of HRM as a method of conducting surveillance when sequencing infrastructures are limited. Given the relatively straightforward assay design, this method could be adapted and applied to a range of pathogens and be tailored for local epidemiological questions outside of SARS-CoV-2 surveillance. For example, HRM has previously been used for screening for bacteria, antimicrobial resistance genes, and Plasmodium falciparum (30–33).
The assays described here are single-tube assays providing results in 3 hours from RNA to variant identification, making them quicker than WGS with far more accessible and streamlined analysis. This technique can make VOC surveillance less costly and more rapid, reducing the wait time from sample to result and reducing reliance and potential overburdening of local and external sequencing infrastructures. This assay’s high sensitivity and specificity have allowed us to investigate the molecular epidemiology of the VOC circulating in Burkina Faso and Kenya during the sample collection windows.
Conclusion
HRM provides a quick, low-cost alternative to sequencing that can provide sensitive and specific identification of key mutations in three of the main VOCs of SARS-CoV-2: Alpha, Delta, and Omicron. We have demonstrated that the assays are flexible, easily updatable, and readily applied to retrospective data sets. The use of these assays would not only reduce the cost of genomic surveillance but prevent overwhelming existing sequencing infrastructure during a pandemic or outbreak situation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1World Health Organization. Tracking SARS-Co V-2 variants. Accessed 9 June 2023. https://www.who.int/activities/tracking-SARS-Co V-2-variants.
- 2Tegally H, San JE, Cotten M, Moir M, Tegomoh B, Mboowa G, Martin DP, Baxter C, Lambisia AW, Diallo A, et al.. 2022. The evolving SARS-Co V-2 epidemic in Africa: Insights from rapidly expanding genomic surveillance. Science 378:eabq 5358. doi:10.1126/science.abq 535836108049 PMC 9529057 · doi ↗ · pubmed ↗
- 3Genomic sequencing effort for SARS-Co V-2 by country during the pandemic. Elsevier Enhanced Reader [Internet]. Available from: https://reader.elsevier.com/reader/sd/pii/S 1201971220325571?token=E 1DF 899CA 6780 DF 91E 7CF 7AA 0F 6375 CDCF 7735 C 66C 96700457 B 0659 F 459E 7459 C 7D 57D 55B 2F 8957863 E 087A 296D 6841 A&origin Region=eu-west-1&origin Creation=20221222153346. Retrieved 2222 Dec December 2022. Accessed , 2222 Dec December 2022
- 4WHO, Regional Office for Africa [Internet].2023. Scaling up genomic sequencing in Africa. Available from: https://www.afro.who.int/news/scaling-genomic-sequencing-africa
- 5Aoki A, Adachi H, Mori Y, Ito M, Sato K, Okuda K, Sakakibara T, Okamoto Y, Jinno H. 2022. Discrimination of SARS-Co V-2 Omicron sublineages BA.1 and BA.2 using a high-resolution melting-based assay: a pilot study. Microbiol Spectr 10:e 01367-22. doi:10.1128/spectrum.01367-2235861527 PMC 9430517 · doi ↗ · pubmed ↗
- 6Moradzad M, Soltani H, Salehi H, Rahmani K, Khateri D, Rahimi MZ, Az D, Fakhari S. 2022. A screening strategy for identifying the dominant variant of SARS-COV-2 in the fifth peak of Kurdistan- Iran population using HRM and Probe-based RT-PCR assay. J Virol Methods 304:114514. doi:10.1016/j.jviromet.2022.11451435271889 PMC 8900876 · doi ↗ · pubmed ↗
- 7Aoki A, Adachi H, Mori Y, Ito M, Sato K, Kinoshita M, Kuriki M, Okuda K, Sakakibara T, Okamoto Y, Jinno H. 2023. A modified high-resolution melting-based assay (HRM) to identify the SARS-Co V-2 N 501Y variant. J Virol Methods 314:114678. doi:10.1016/j.jviromet.2023.11467836681163 PMC 9847321 · doi ↗ · pubmed ↗
- 8Aoki A, Adachi H, Mori Y, Ito M, Sato K, Okuda K, Sakakibara T, Okamoto Y, Jinno H. 2021. A rapid screening assay for L 452R and T 478K spike mutations in SARS-Co V-2 Delta variant using high-resolution melting analysis. J Toxicol Sci 46:471–476. doi:10.2131/jts.46.47134602531 · doi ↗ · pubmed ↗