Synthesis and Reactivity of Lewis-Base-Supported Terminal Thorium Imido Metallocene, (η5‑C5Me5)2ThN(p‑tolyl)(dmap)2
Yi Heng, Enwei Zhou, Dongwei Wang, Wanjian Ding, Guohua Hou, Guofu Zi, Marc D. Walter

TL;DR
Scientists synthesized a thorium imido metallocene and found it can participate in various chemical reactions, including cycloadditions and deprotonation.
Contribution
The synthesis and reactivity of a Lewis base-supported terminal thorium imido metallocene are reported, showcasing diverse chemical transformations.
Findings
The thorium imido metallocene undergoes multiple cycloaddition reactions with sulfur, selenium, alkynes, and other organic compounds.
The imido group acts as a nucleophile toward metal halides, esters, and azidosilanes.
Substituent effects on ligands influence the reactivity of thorium imido metallocenes.
Abstract
Treatment of (η5-C5Me5)2ThMe2 (1) with p-tolylNH2 in toluene, in the presence of 4-dimethylaminopyridine (dmap), affords a Lewis base-supported terminal thorium imido metallocene, (η5-C5Me5)2ThN(p-tolyl)(dmap)2 (5), alongside the release of methane. In toluene solution, an equilibrium is established among complex 5, dmap, and the amido pyridyl complex (η5-C5Me5)2Th[NH(p-tolyl)][κ2-C,N-4-(Me2N)C5H3N] (5′), setting the stage for diverse reactivity. Complex 5 may initiate [2 + 2], [2 + 4], [2 + 1], or [2 + 3] cycloadditions with elemental sulfur and selenium, alkynes, carbodiimides, ketones, thio-ketones, isothiocyanates, CS2, organic nitriles and isonitriles, as well as organic azides. Moreover, imido moiety can act as a nucleophile toward metal halides, esters, and azidosilanes; and it may promote deprotonation reactions with 1-methylimidazole, 2,6-Me2C5H3NO, Me3PO, silanes,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1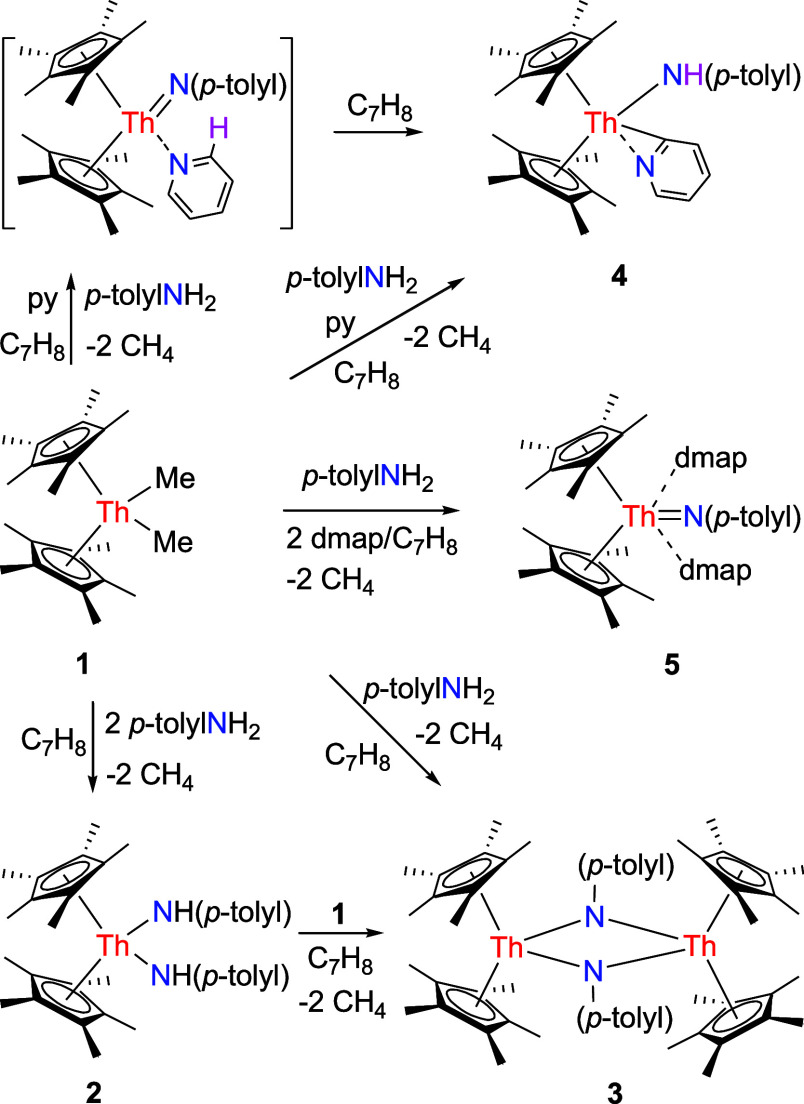 1
1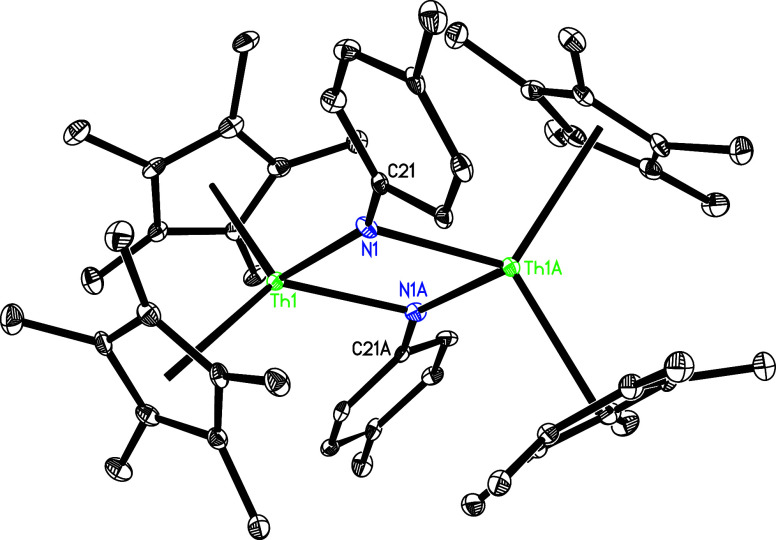 2
2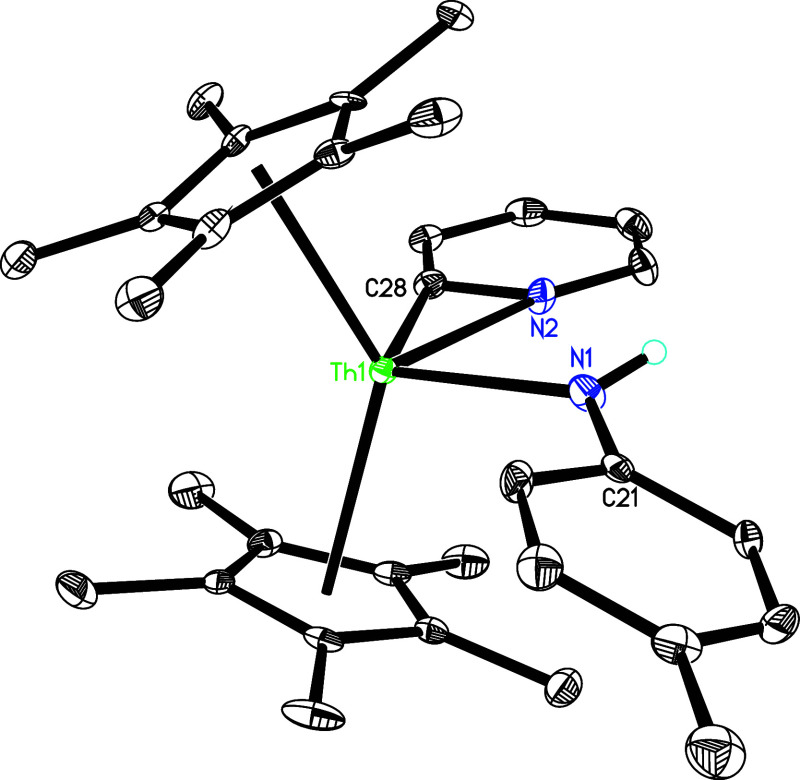 3
3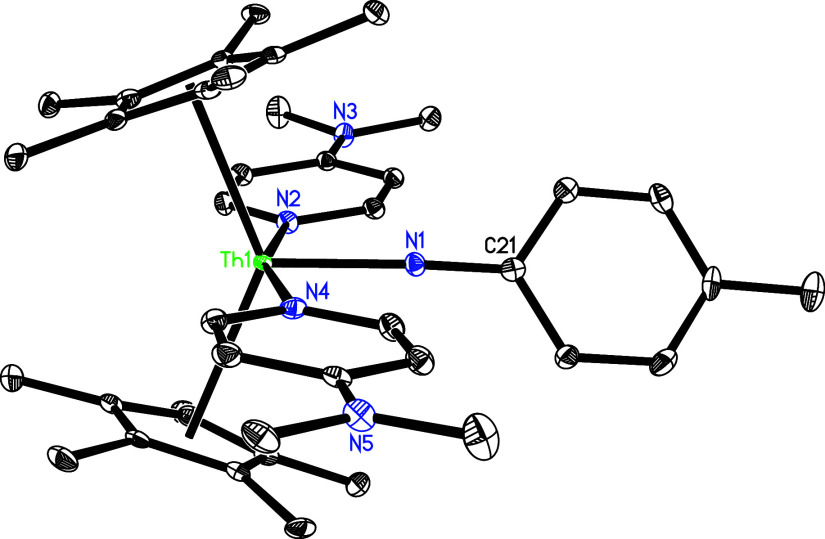 4
4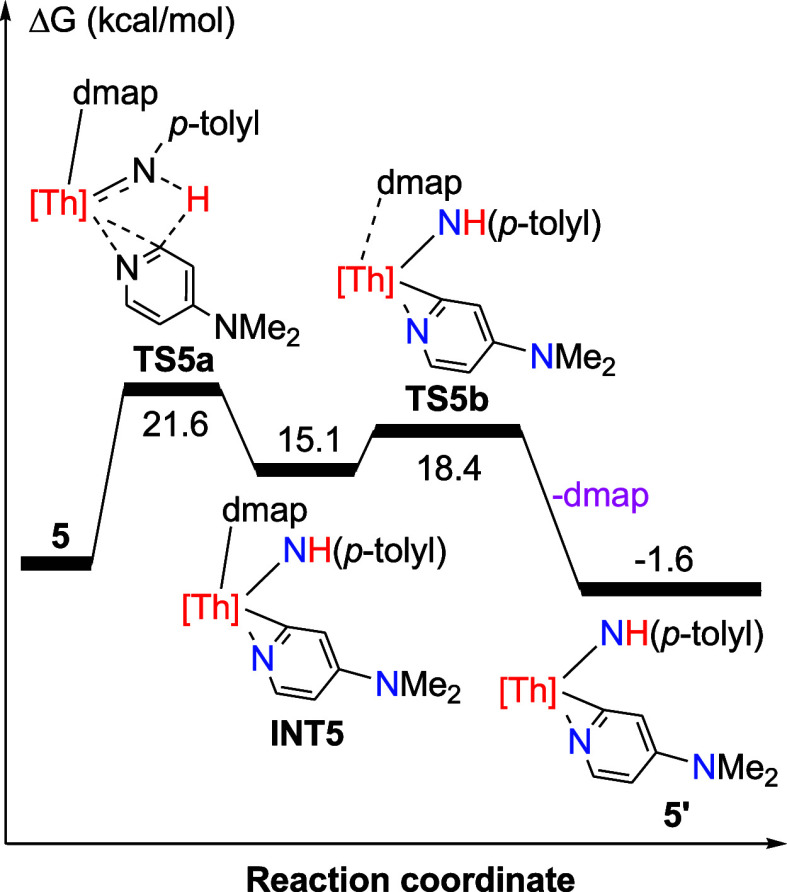 5
5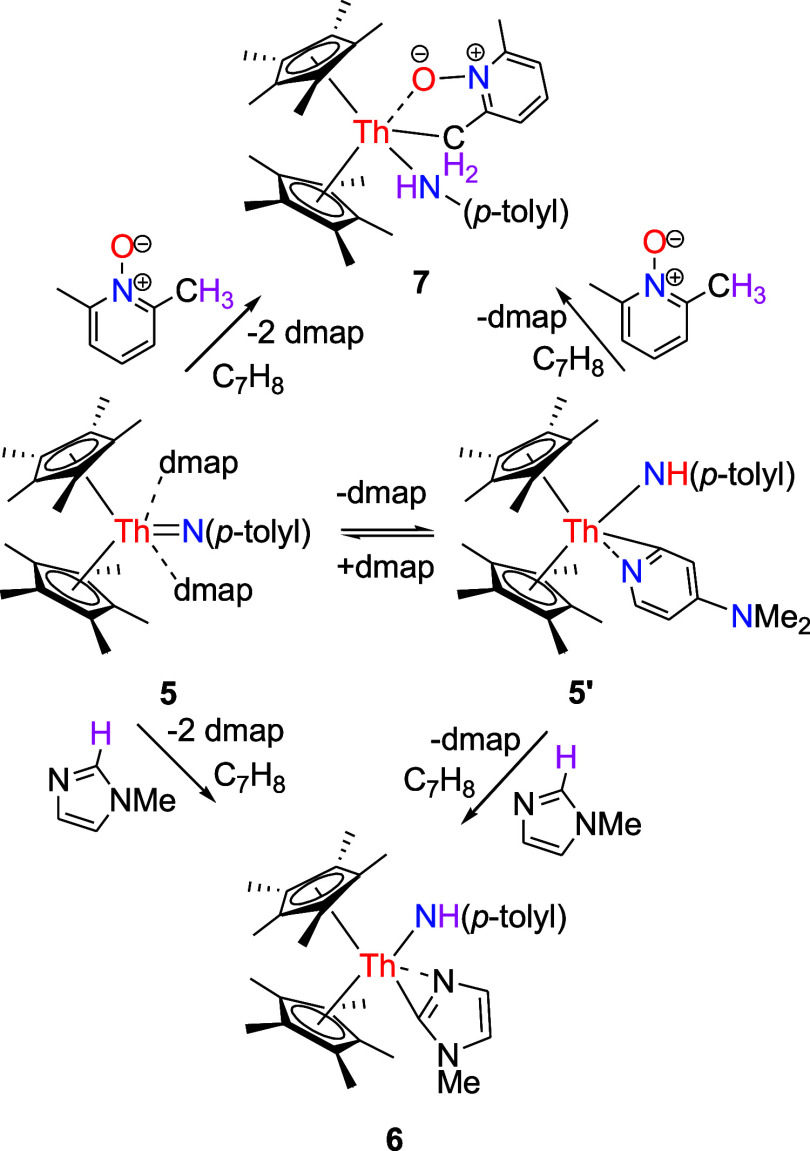 2
2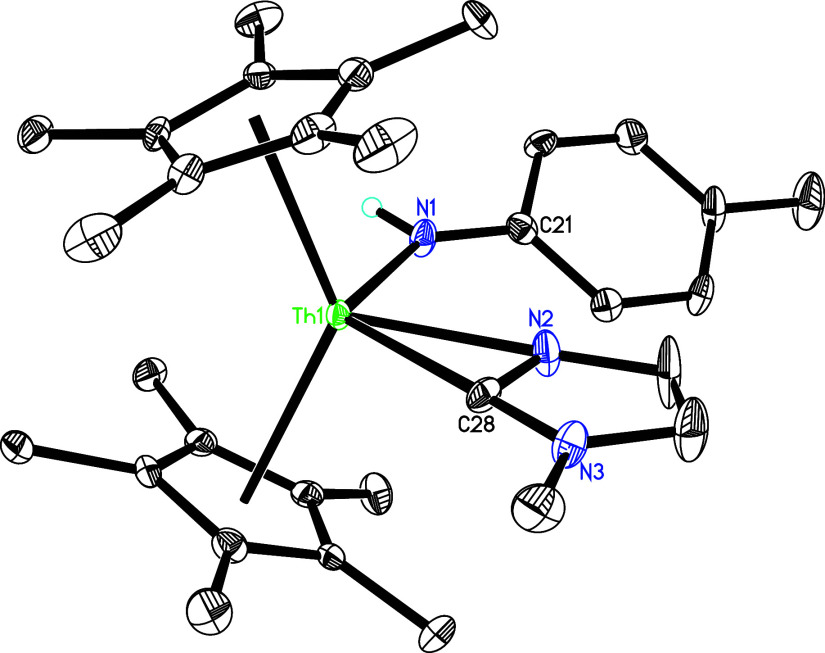 6
6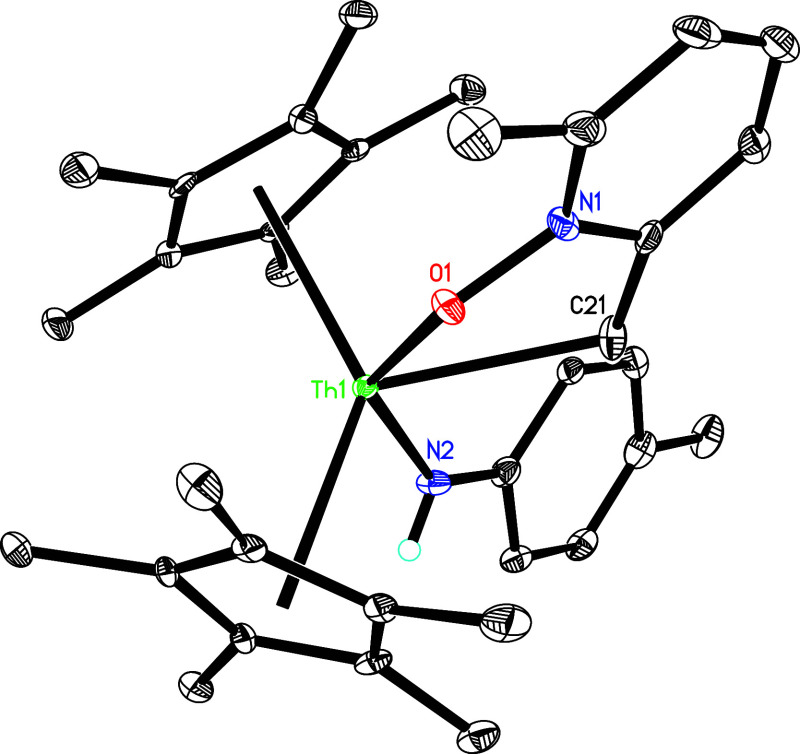 7
7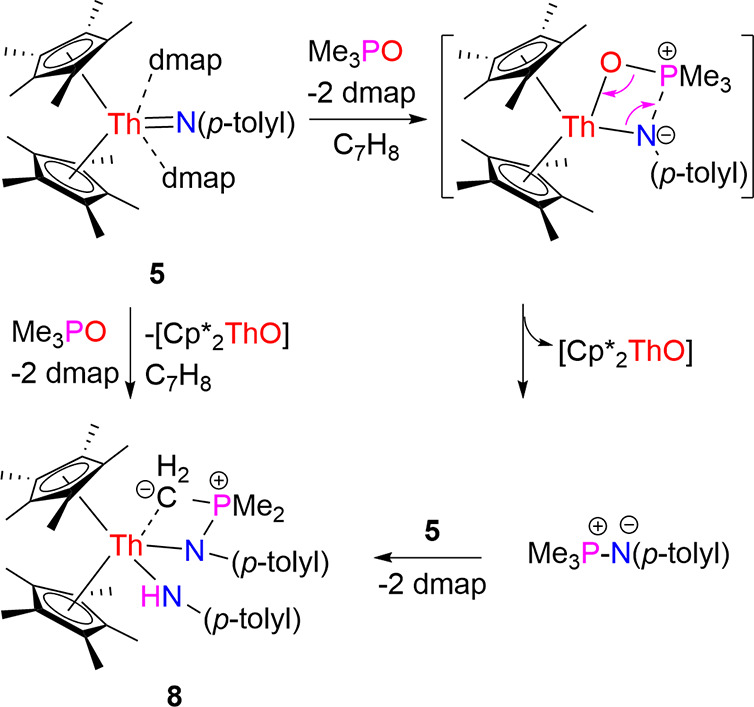 3
3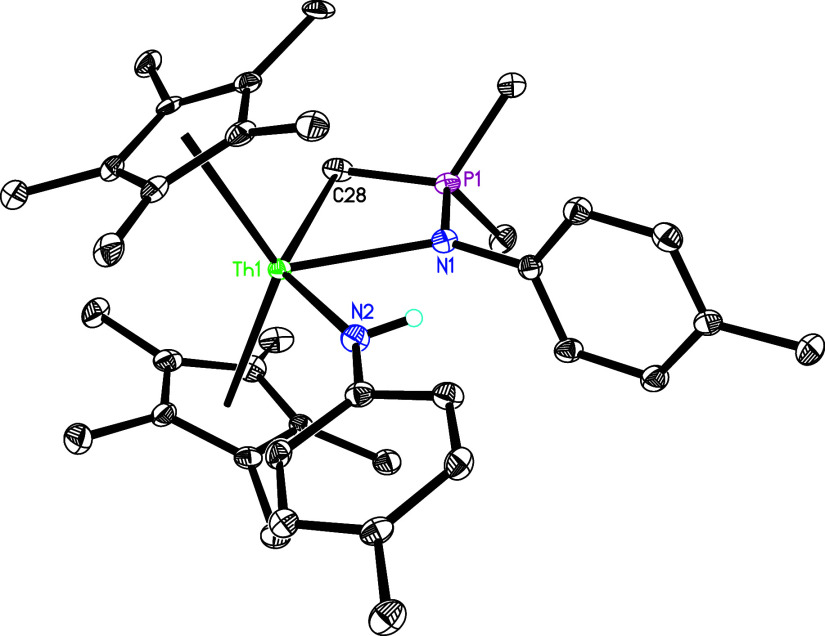 8
8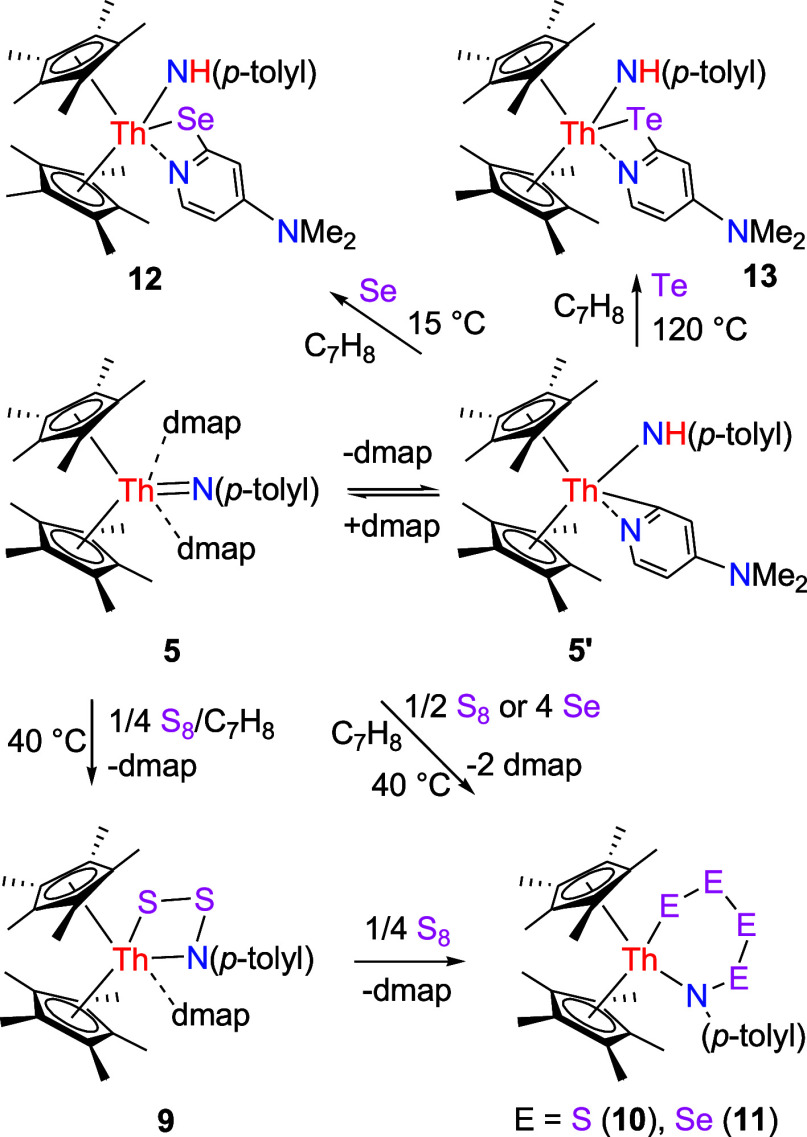 4
4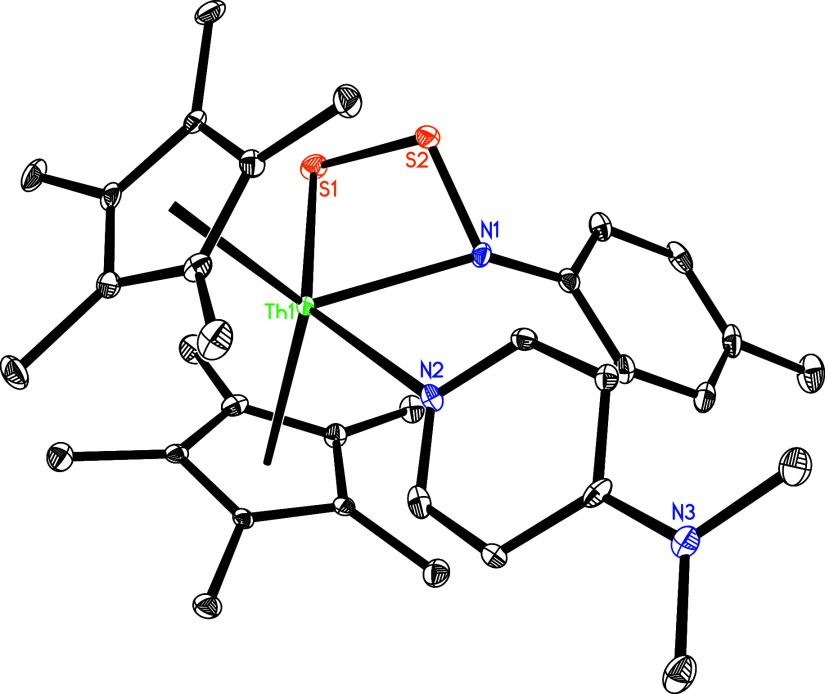 9
9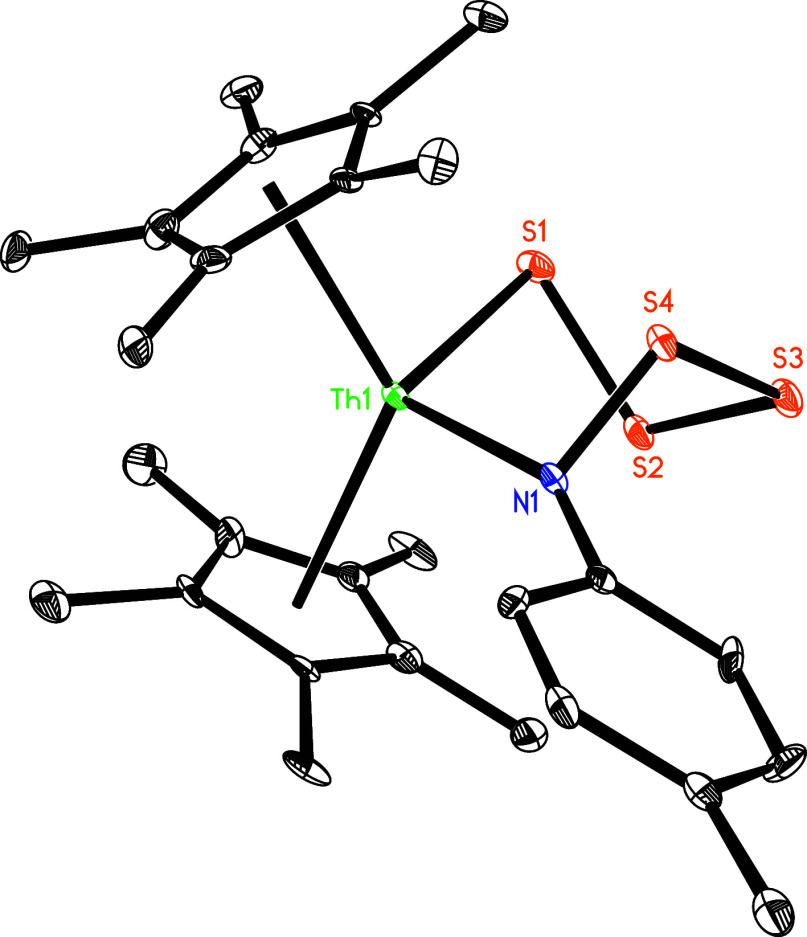 10
10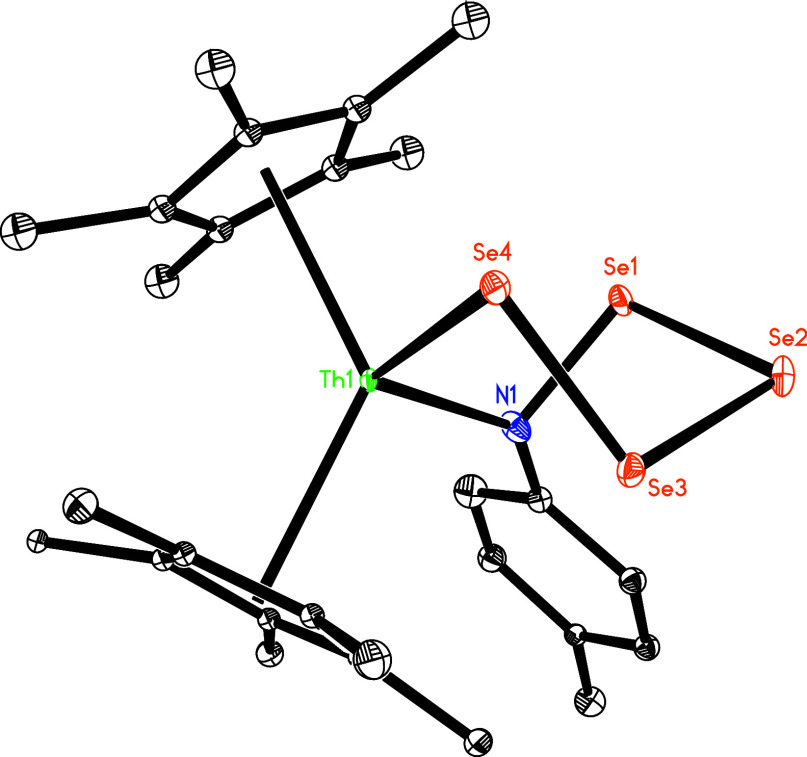 11
11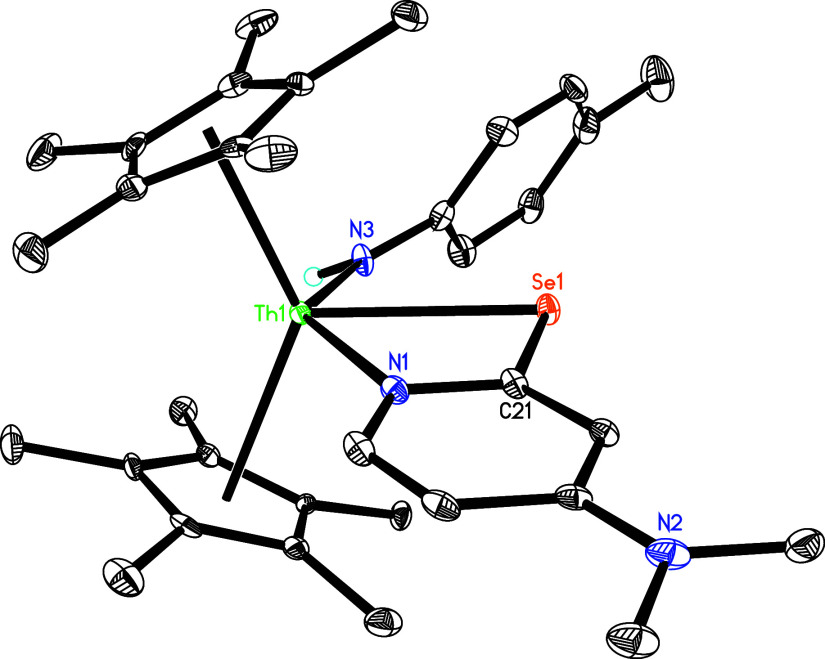 12
12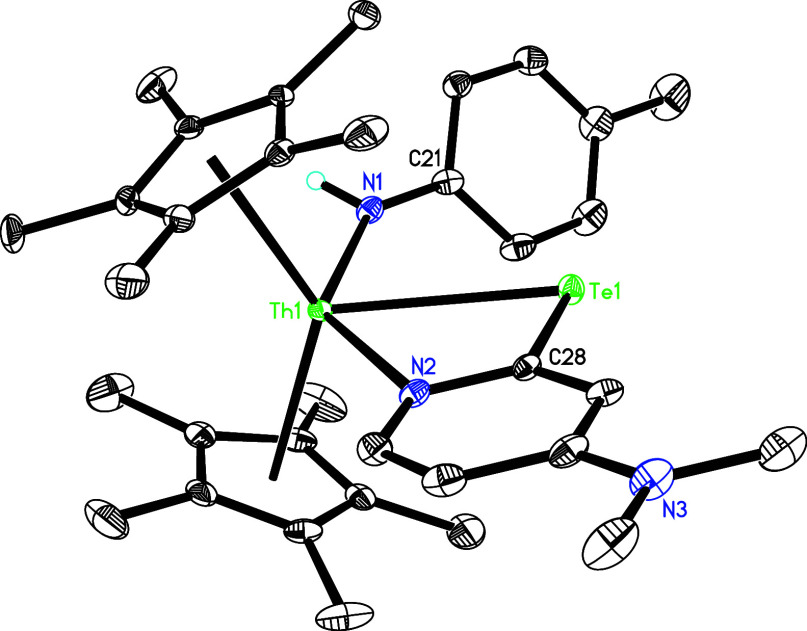 13
13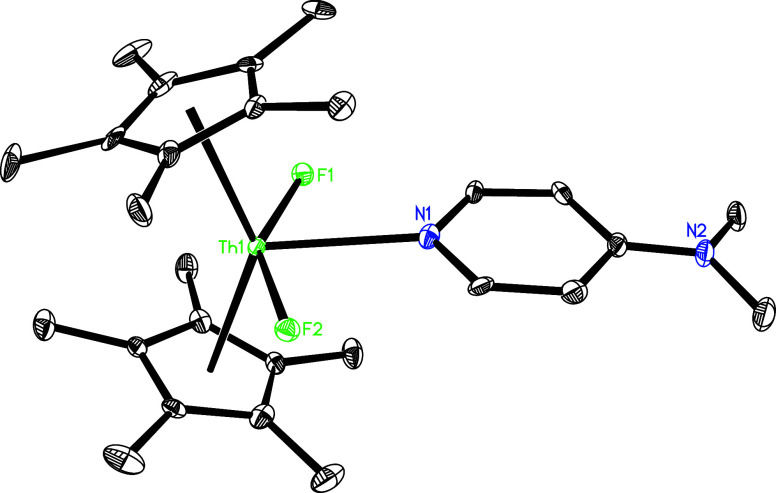 14
14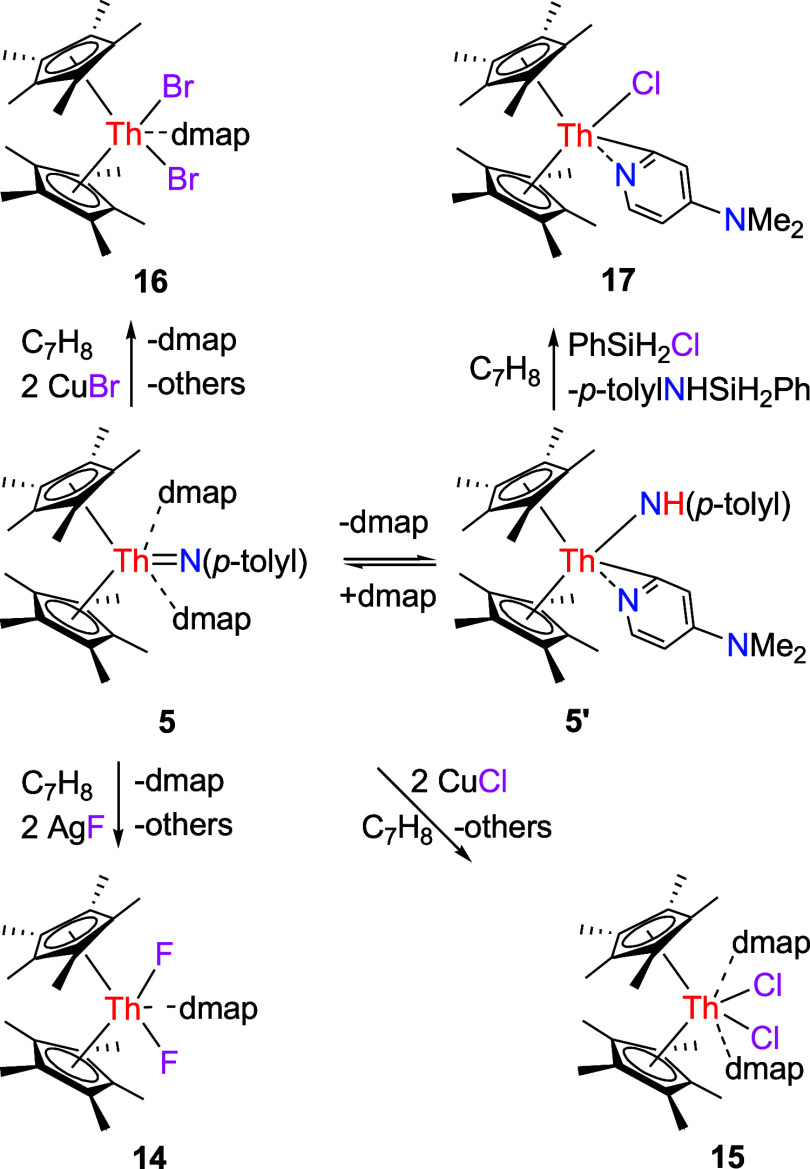 5
5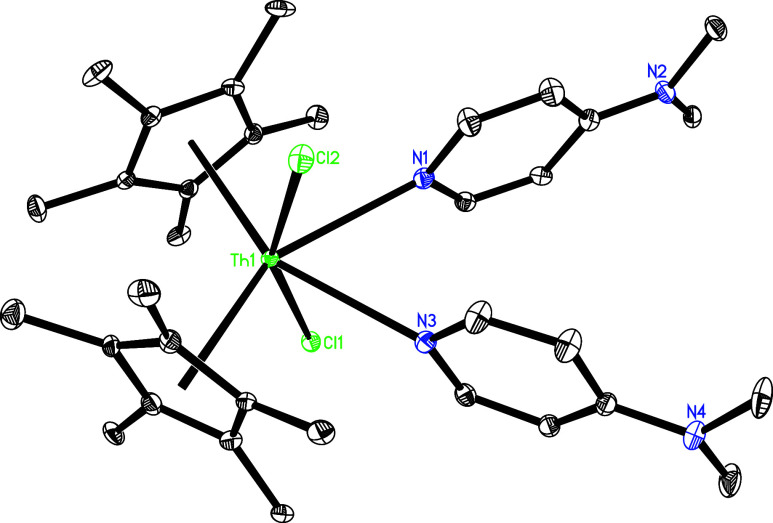 15
15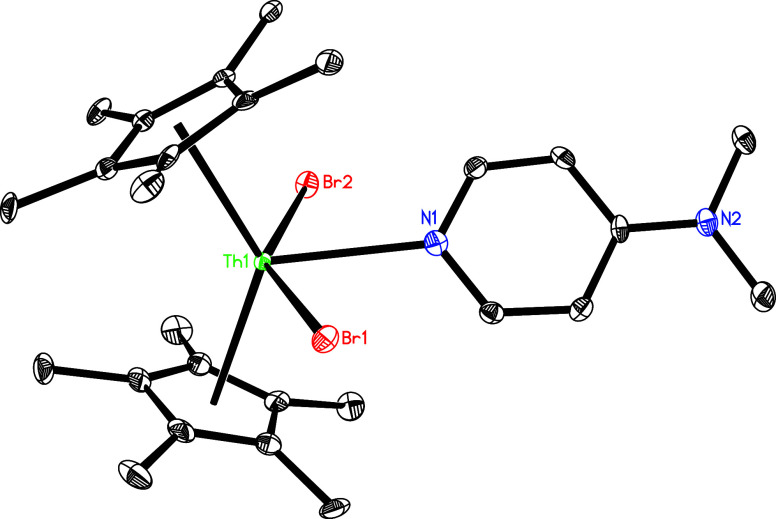 16
16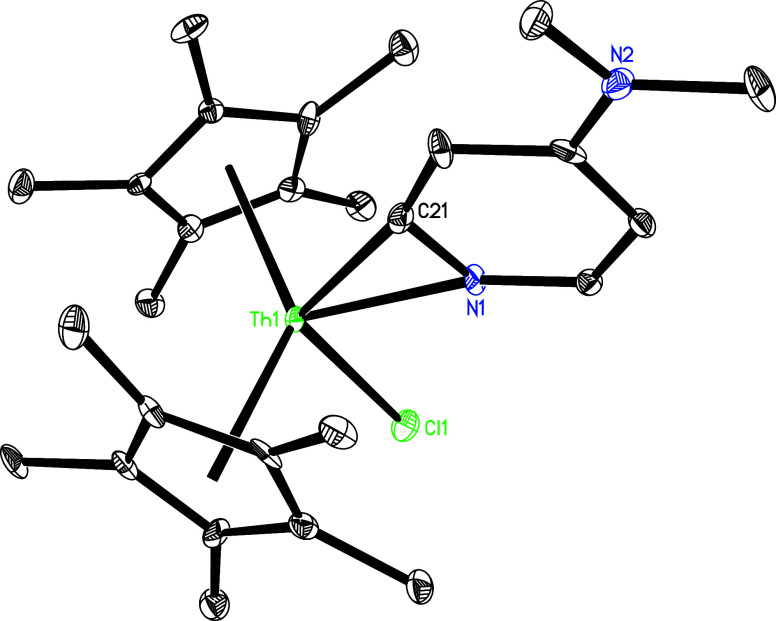 17
17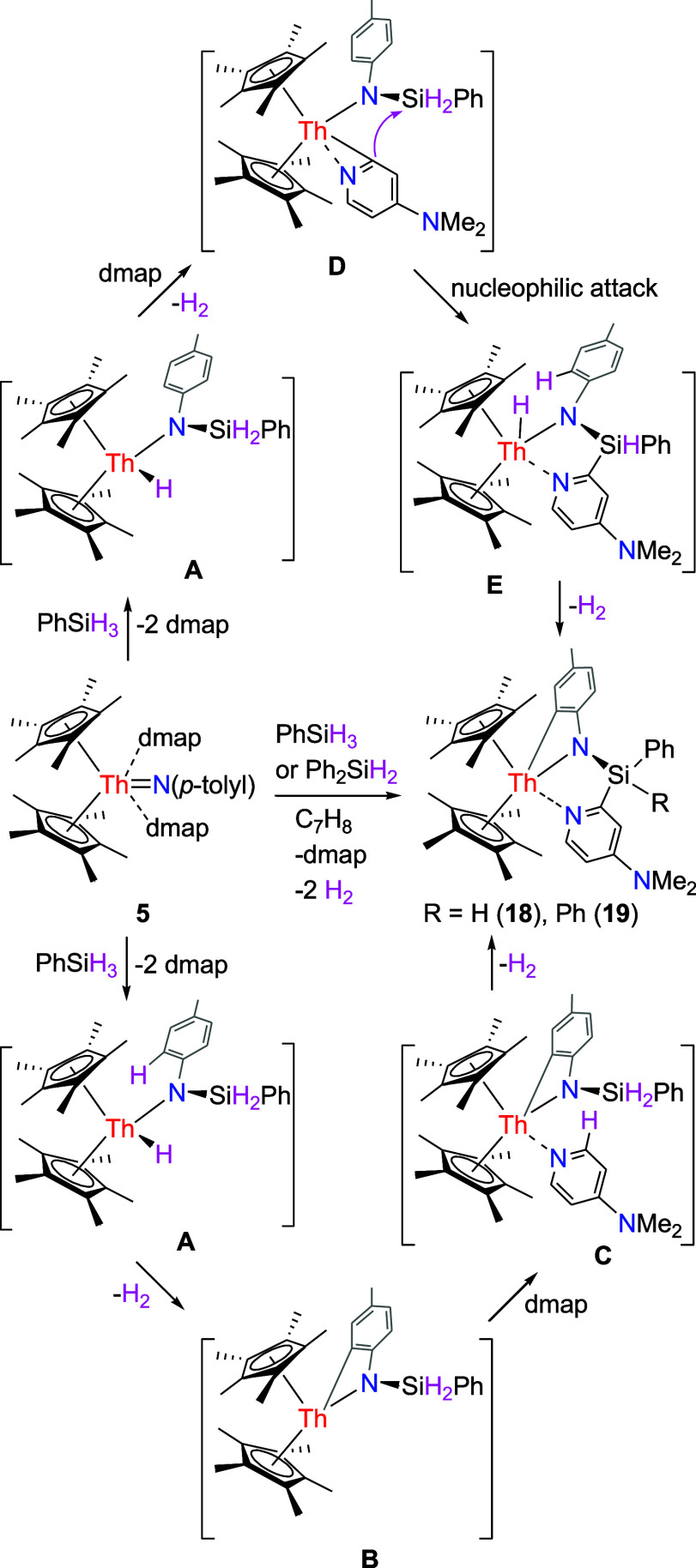 6
6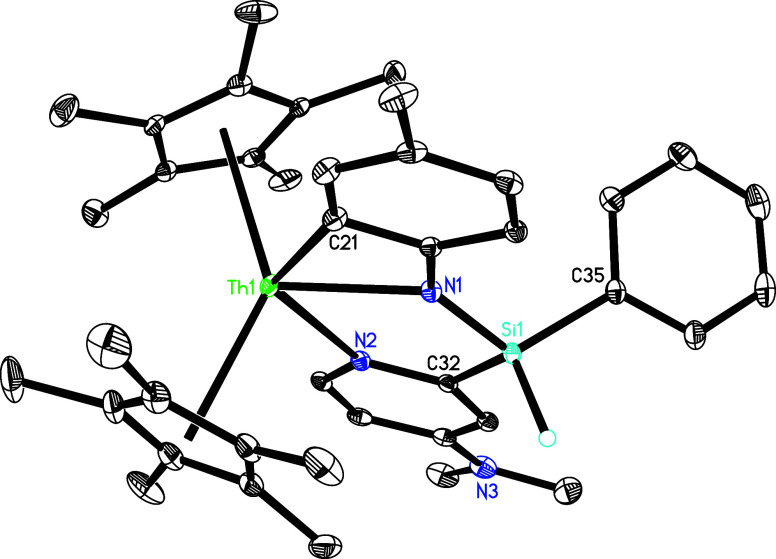 18
18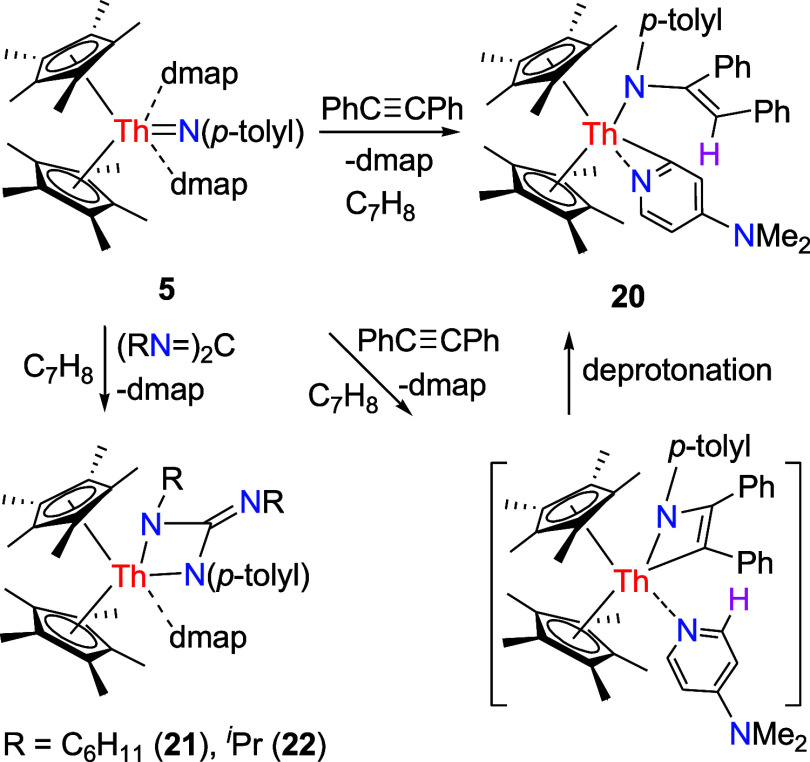 7
7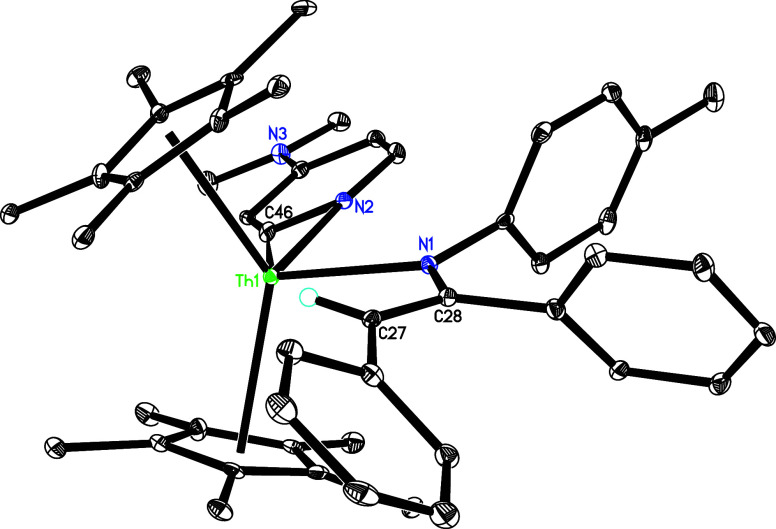 19
19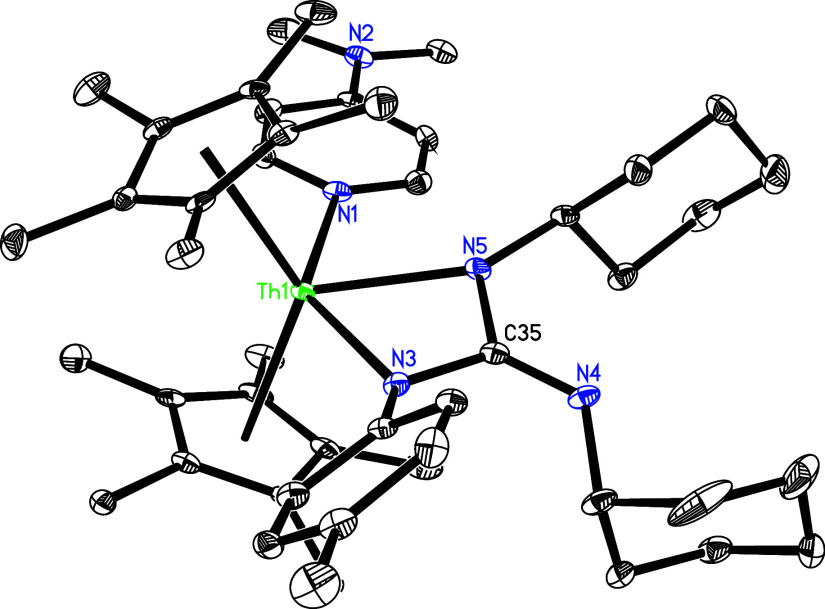 20
20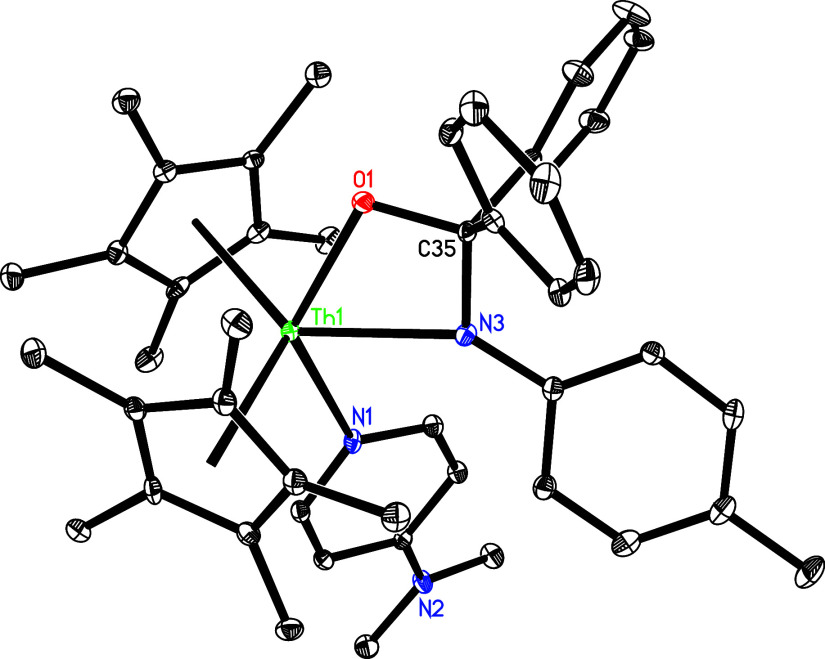 21
21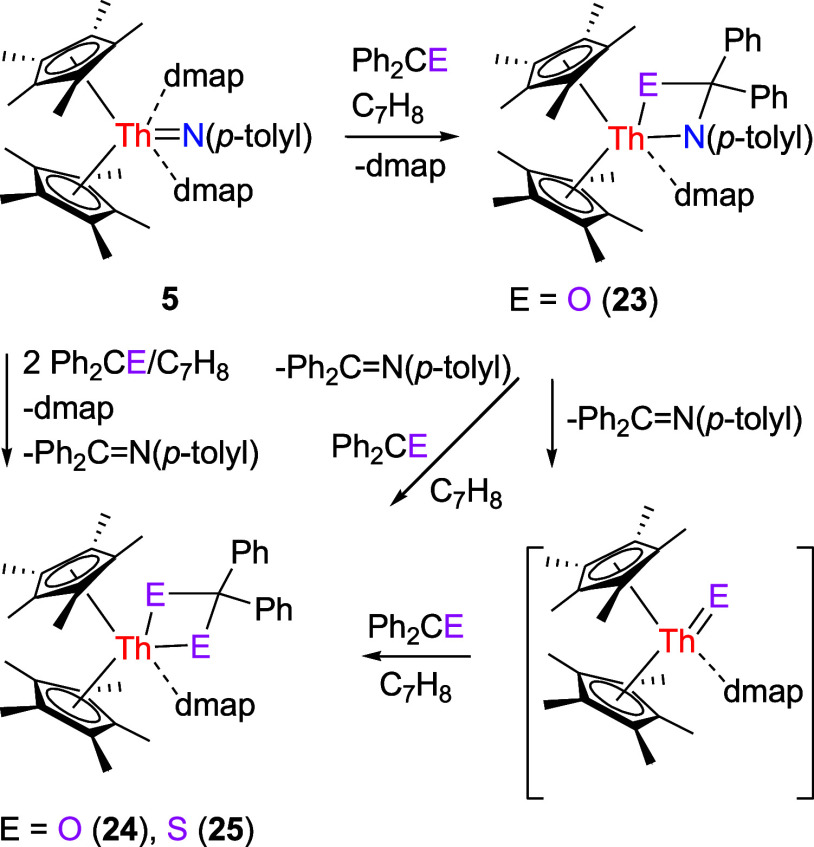 8
8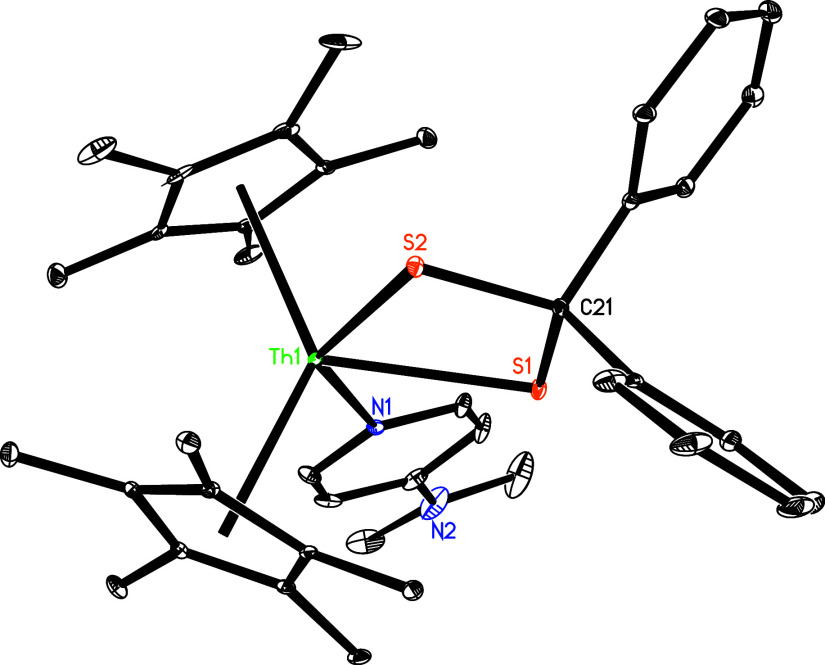 22
22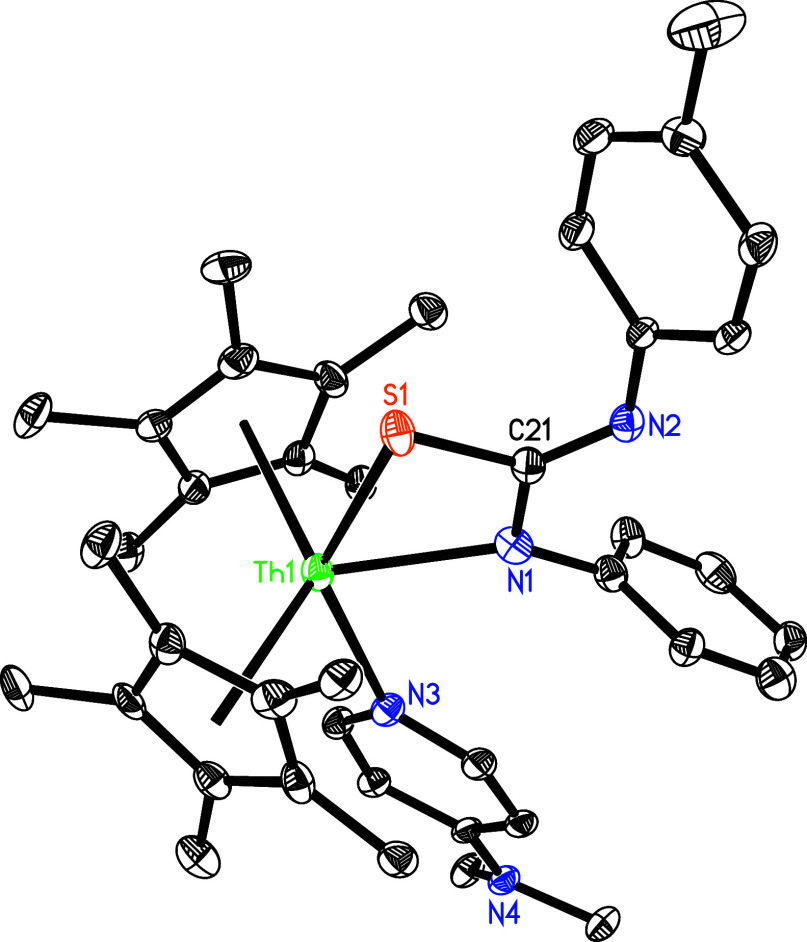 23
23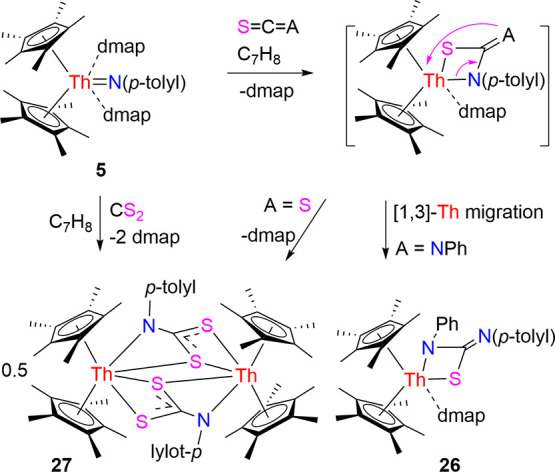 9
9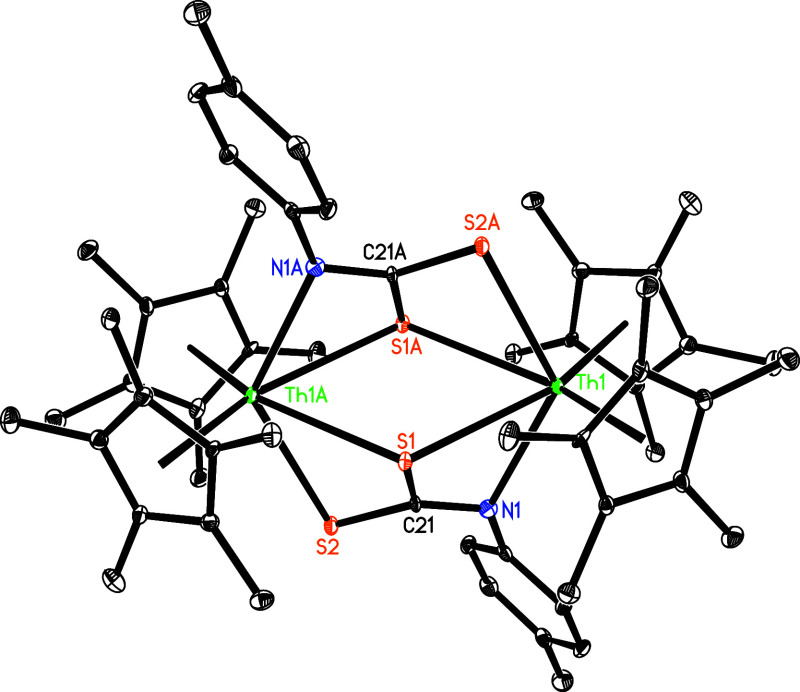 24
24 10
10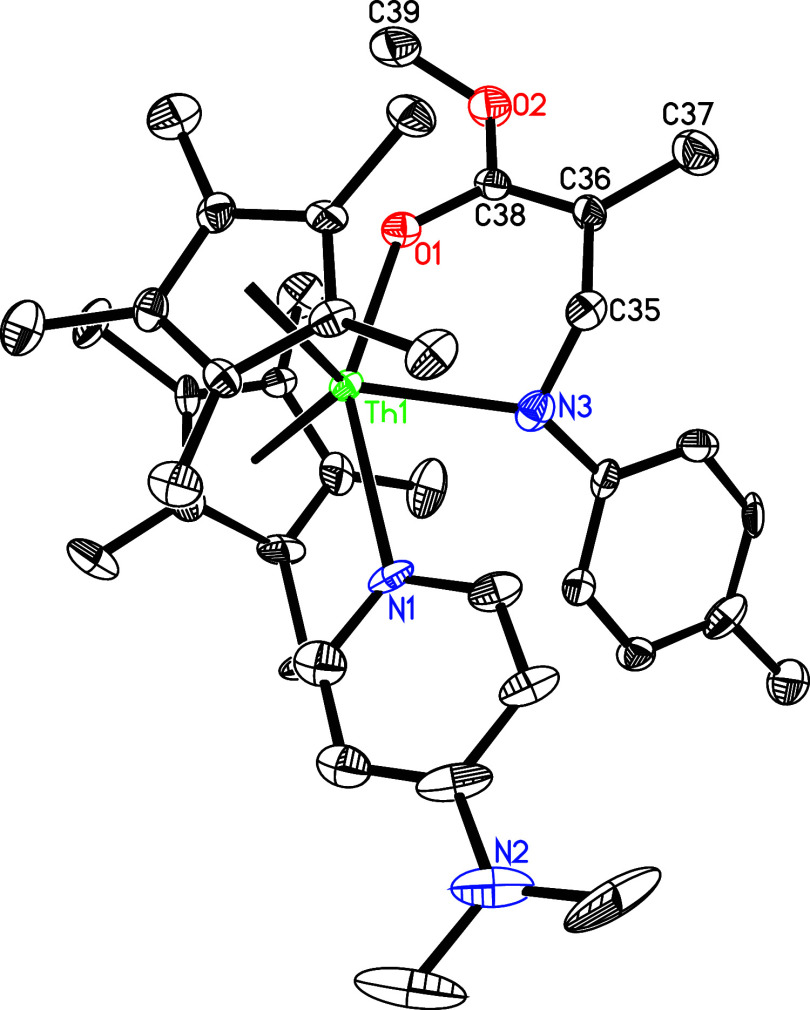 25
25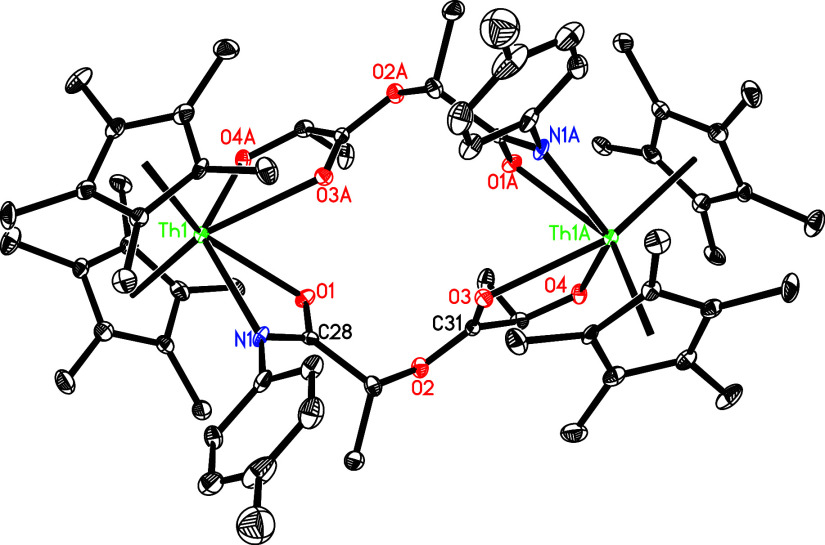 26
26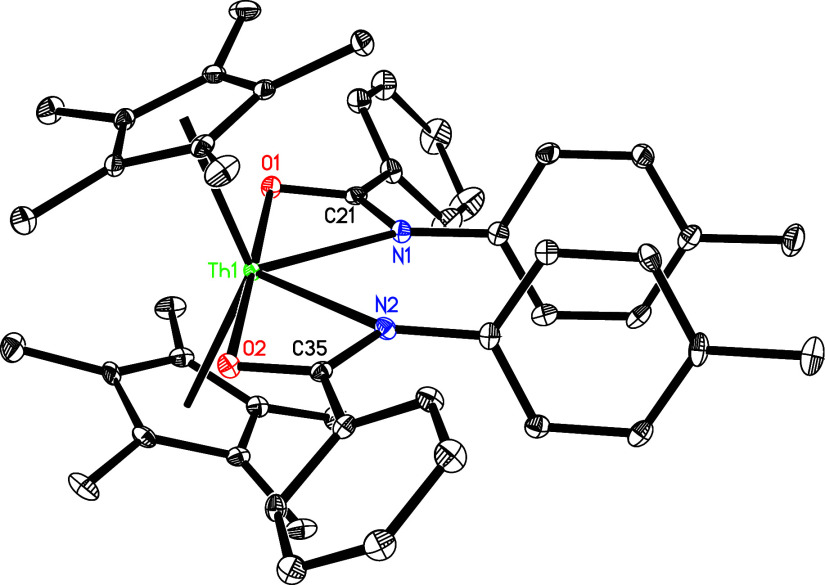 27
27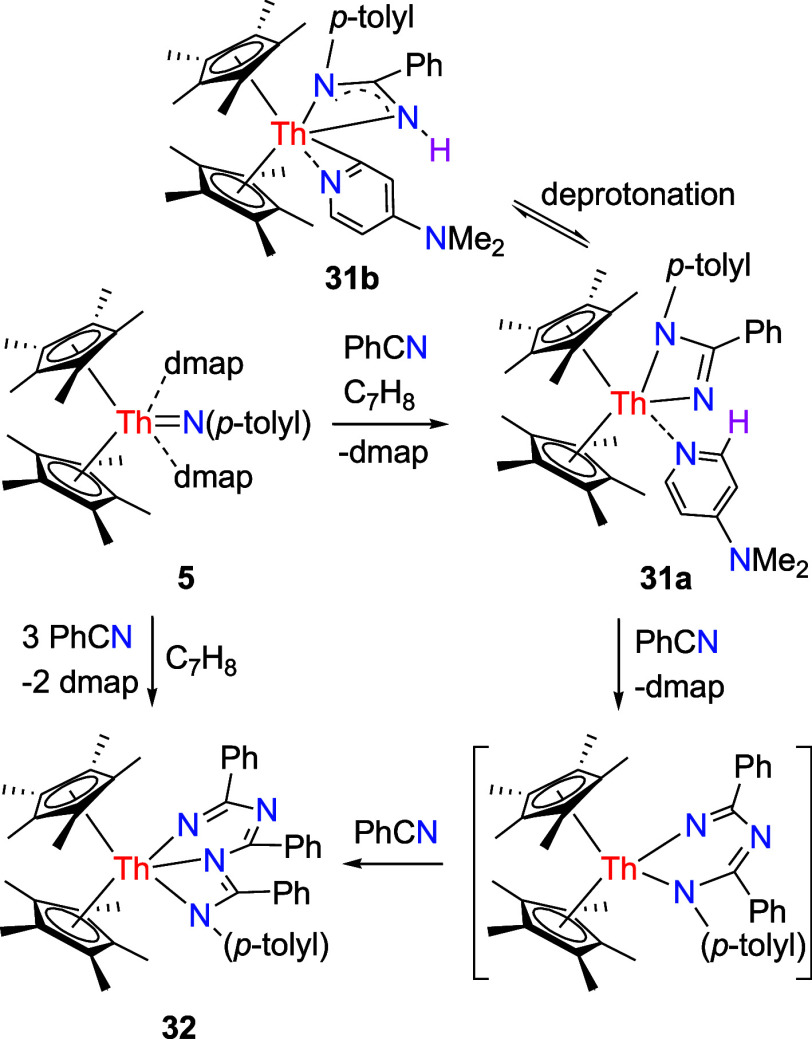 11
11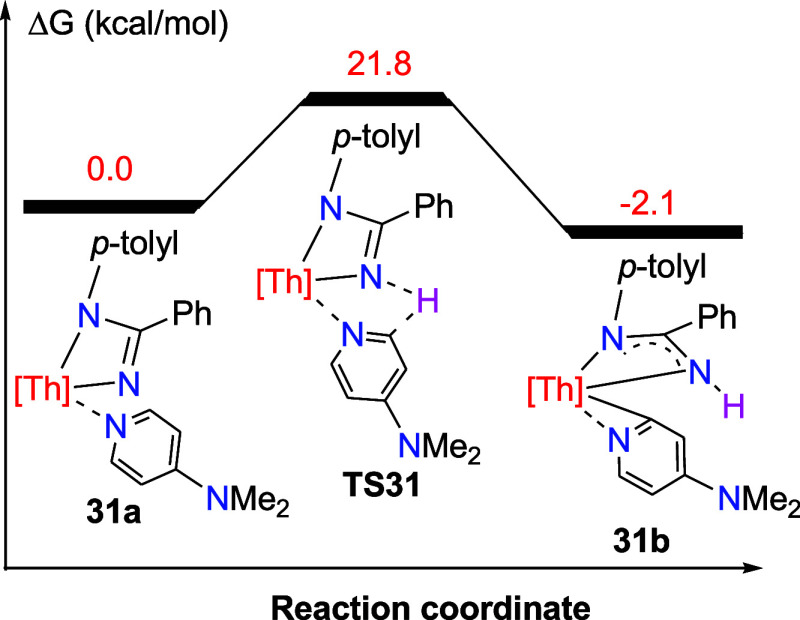 28
28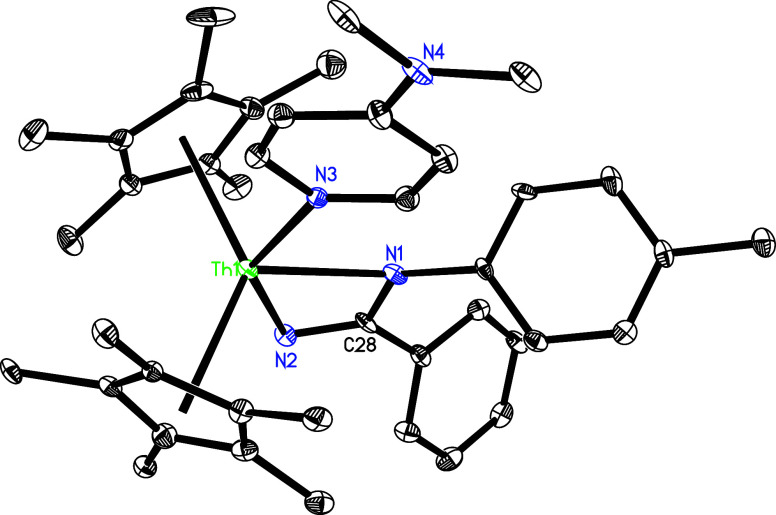 29
29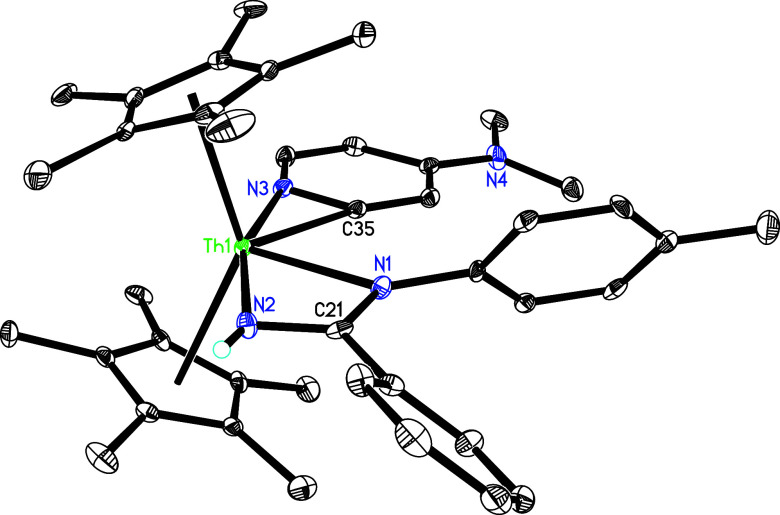 30
30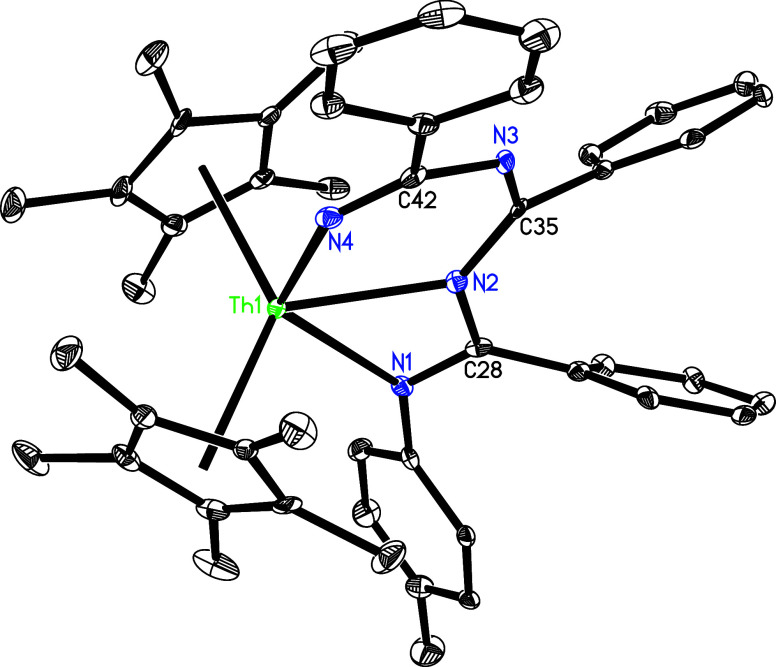 31
31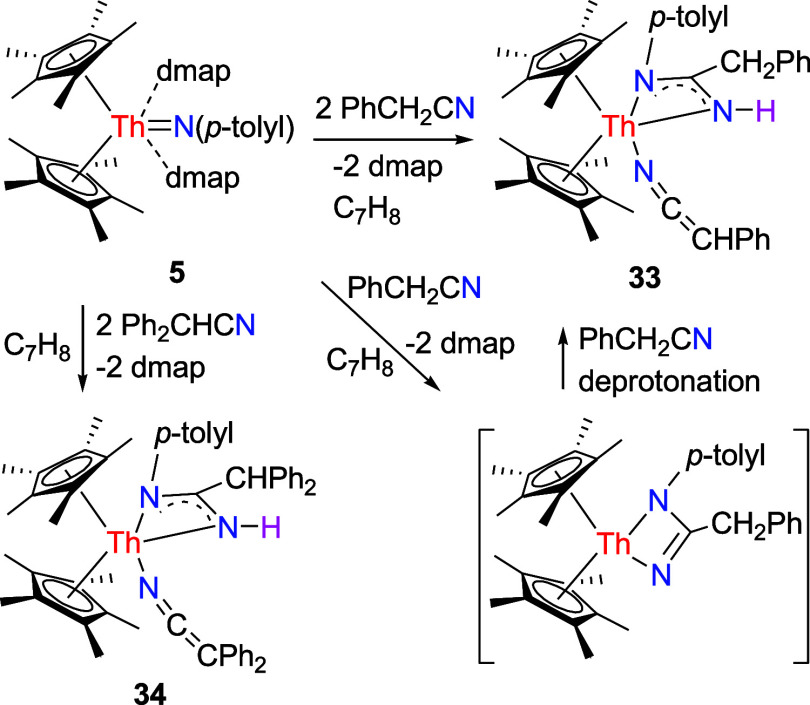 12
12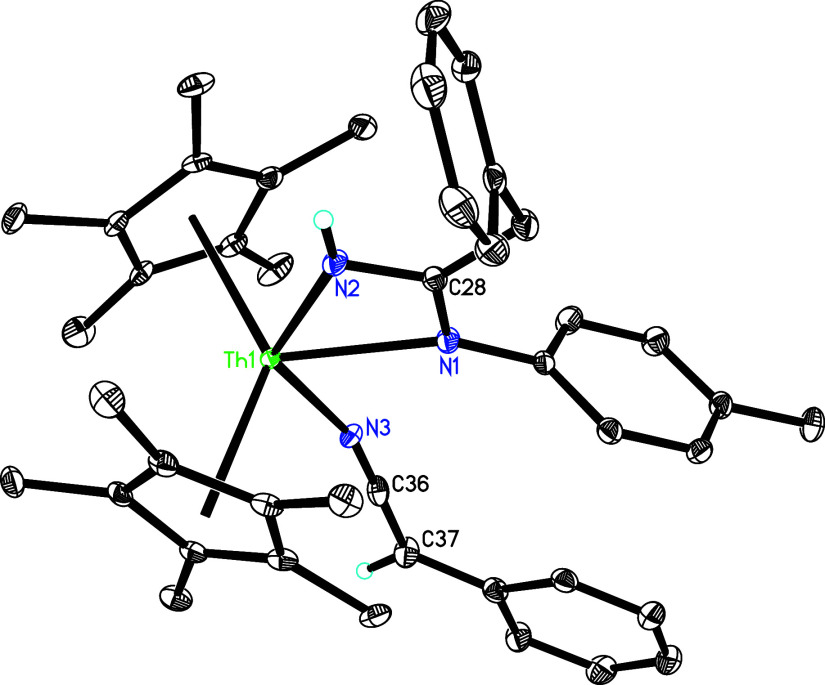 32
32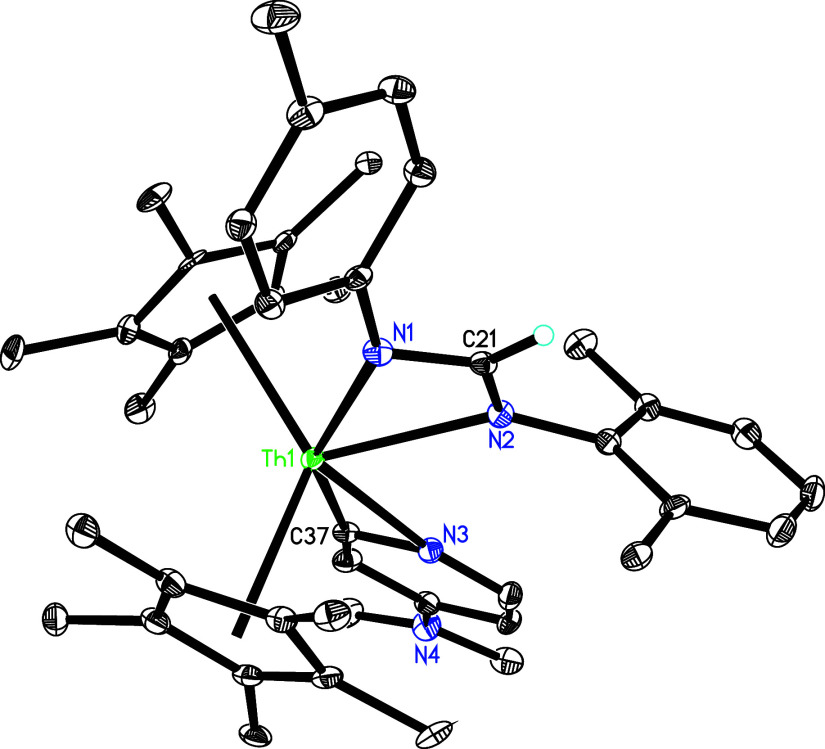 33
33 13
13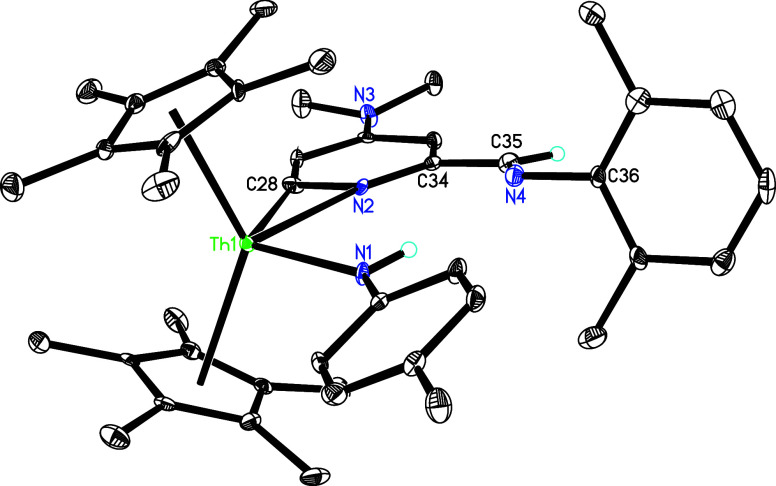 34
34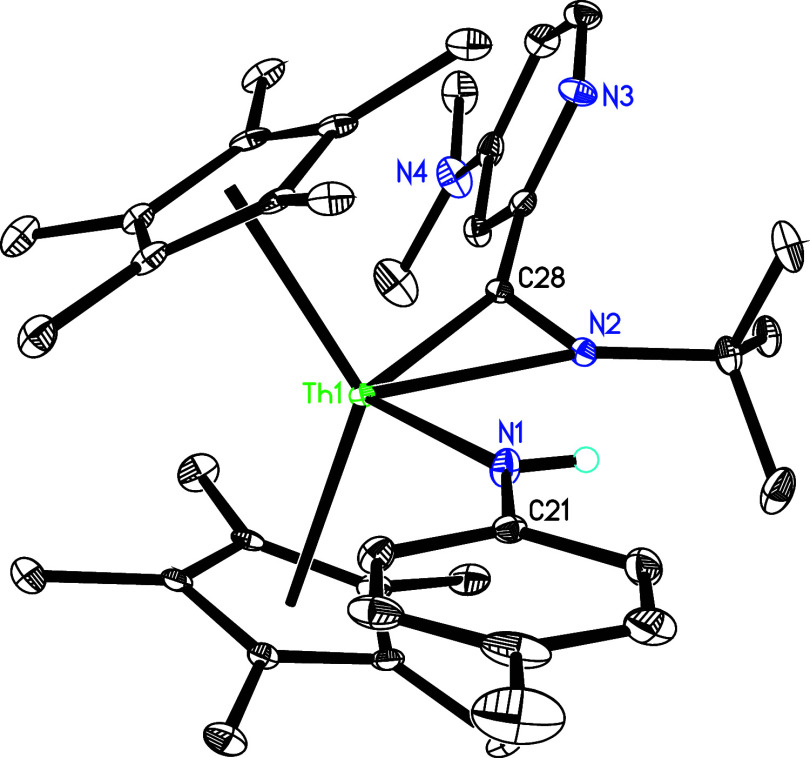 35
35 14
14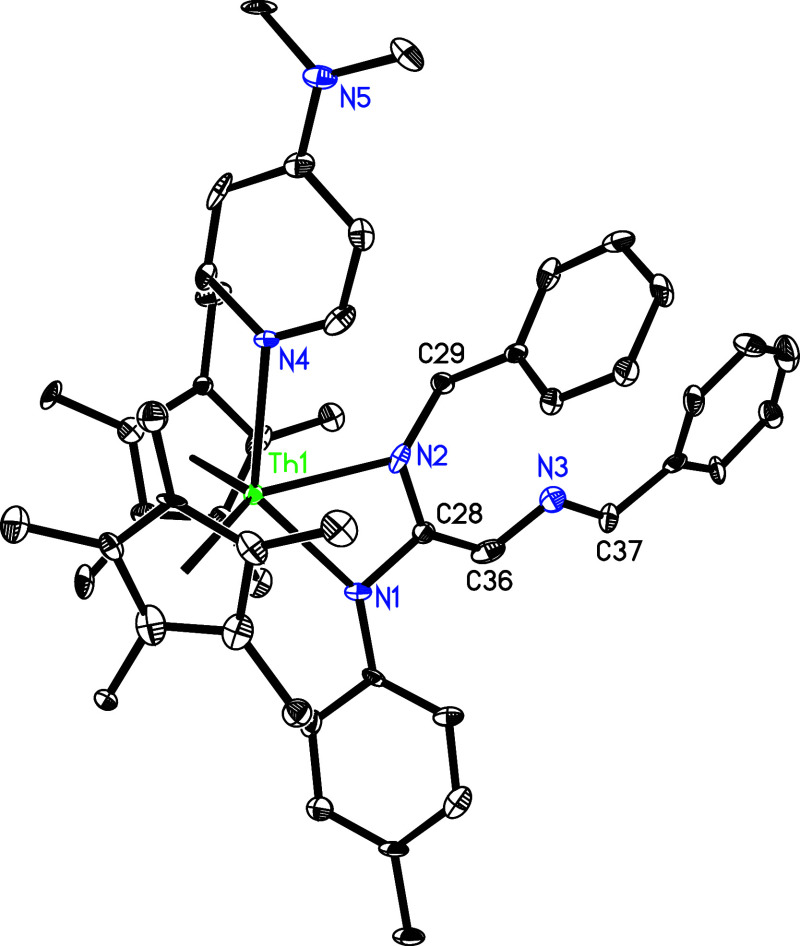 36
36 15
15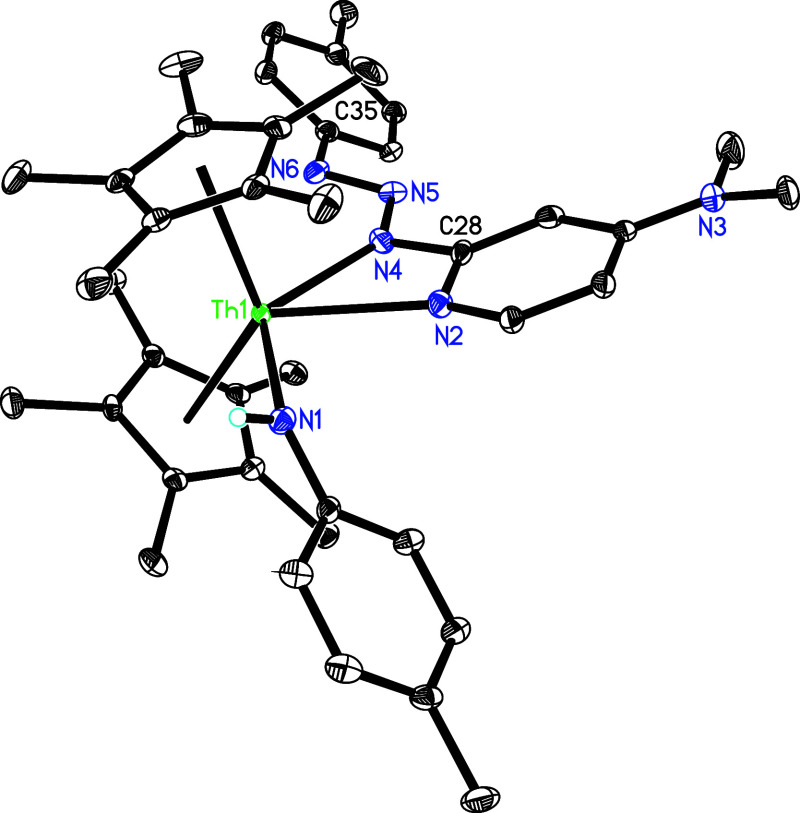 37
37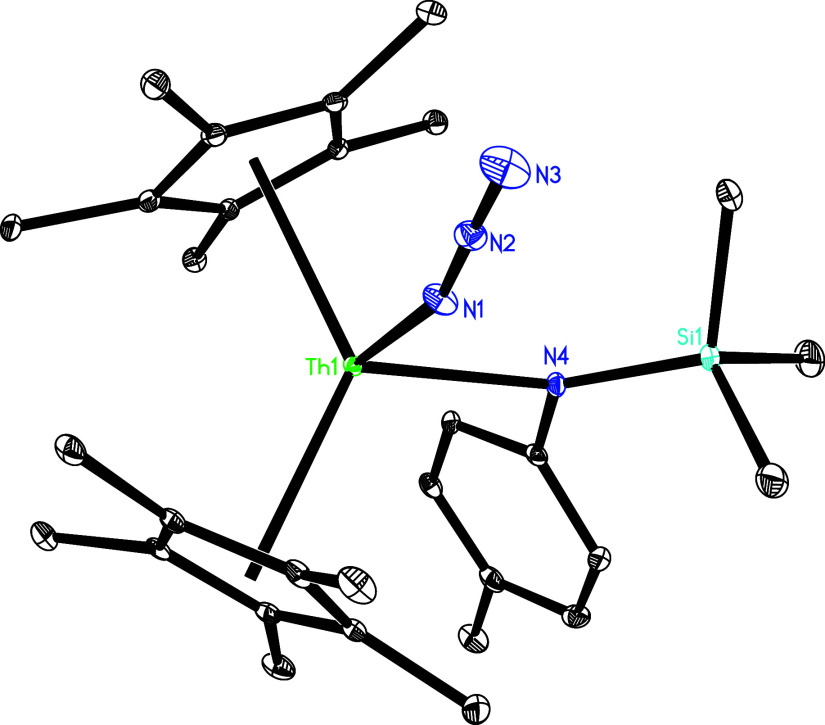 38
38 16
16| compound | C(Cp)-Th | C(Cp)-Th | Cp(cent)-Th | Th-X | Cp(cent)-Th-Cp(cent) | X-Th-X/Y |
|---|---|---|---|---|---|---|
|
| 2.813(4) | 2.797(4)–2.826(4) | 2.539(4), 2.539(4) | N (1) 2.329(3), N(1A) 2.329(3) | 133.3(2) | 103.5(2) |
|
| Th (1) 2.909(17) | Th (1) 2.859(6)–3.002(5) | Th (1) 2.641(5), 2.653(5) | Th (1) N (1) 2.271(5), N(1A) 2.401(5) | Th (1) 117.3(2) | Th (1)
71.4(2) |
|
| 2.828(7) | 2.806(6)–2.862(6) | 2.570(6), 2.545(6) | N (1) 2.355(5), N(2) 2.475(6), C(28) 2.454(7) | 136.2(2) | 31.7(2) |
|
| 2.926(14) | 2.869(4)–2.995(4) | 2.673(4), 2.660(4) | N (1) 2.082(3), N(2) 2.673(3), N(4) 2.634(3) | 135.4(1) | 164.1(1) |
|
| 2.842(12) | 2.786(7)–2.900(7) | 2.591(9), 2.581(9) | N (1) 2.365(7), N(2) 2.448(7), C(28) 2.485(7) | 133.1(2) | 31.7(2) |
|
| 2.859(12) | 2.802(7)–2.900(8) | 2.603(7), 2.574(7) | N(2) 2.351(6), O (1) 2.418(5), C(21) 2.598(8) | 134.9(2) | 62.6(2) |
|
| 2.867(9) | 2.829(6)–2.908(6) | 2.599(6), 2.597(6) | N (1) 2.489(6), N(2) 2.348(5), C(28) 2.700(7) | 130.5(2) | 60.4(2) |
|
| 2.854(7) | 2.827(4)–2.886(5) | 2.588(4), 2.586(4) | N (1) 2.402(4), N(2) 2.661(4), S (1) 2.886(1) | 127.4(1) | 68.1(1) |
|
| 2.826(2) | 2.815(12)–2.836(10) | 2.552(10), 2.558(10) | N (1) 2.380(11), S (1) 2.819(4) | 128.7(3) | 109.8(3) |
|
| 2.829(8) | 2.794(9)–2.853(9) | 2.560(9), 2.568(9) | N (1) 2.351(8), Se (1) 2.927(1) | 128.3(3) | 113.3(2) |
|
| 2.842(11) | 2.798(5)–2.881(5) | 2.565(5), 2.582(5) | N (1) 2.590(4), N(3) 2.348(4), Se (1) 2.959(1) | 134.4(5) | 59.2(2) |
|
| 2.832(11) | 2.778(6)–2.881(6) | 2.576(6), 2.556(6) | N (1) 2.333(5), N(2) 2.594(5), Te (1) 3.162(2) | 134.8(2) | 60.3(2) |
|
| 2.818(7) | 2.789(10)–2.852(9) | 2.550(9), 2.545(9) | N (1) 2.607(6), F (1) 2.182(5), F(2) 2.184(5) | 140.7(3) | 149.0(2) |
|
| 2.888(7) | 2.851(10)–2.911(10) | 2.621(10), 2.628(10) | Cl (1) 2.732(1), Cl(2) 2.735(1), N (1) 2.746(4), N(3) 2.737(4) | 121.4(3) | 156.6(1) |
|
| 2.826(6) | 2.791(6)–2.847(5) | 2.564(6), 2.545(6) | N (1) 2.628(5), Br (1) 2.872(1), Br(2) 2.921(1) | 133.4(2) | 148.1(1) |
|
| 2.781(4) | 2.767(10)–2.801(9) | 2.504(9), 2.513(9) | N (1) 2.407(8), Cl (1) 2.678(3), C(21) 2.428(10) | 137.7(3) | 32.2(3) |
|
| 2.871(19) | 2.783(6)–2.936(15) | 2.567(6), 2.658(6) | N (1) 2.356(4), N(2) 2.630(4), C(21) 2.514(5) | 137.8(6) | 69.2(1) |
|
| 2.837(12) | 2.793(3)–2.884(3) | 2.572(3), 2.567(3) | N (1) 2.363(3), N(2) 2.624(3), C(21) 2.488(4) | 135.6(1) | 68.8(1) |
|
| 2.855(9) | 2.813(8)–2.891(8) | 2.590(7), 2.581(7) | N (1) 2.425(6), N(2) 2.432(6), C(46) 2.455(7) | 133.9(2) | 32.1(2) |
|
| 2.864(13) | 2.807(4)–2.912(4) | 2.601(4), 2.599(4) | N (1) 2.658(4), N(3) 2.377(4), N(5) 2.373(4) | 132.6(1) | 57.0(1) |
|
| 2.873(13) | 2.814(6)–2.921(7) | 2.606(7), 2.610(7) | N (1) 2.613(6), N(3) 2.365(5), N(5) 2.344(6) | 130.8(2) | 57.0(2) |
|
| 2.887(15) | 2.816(4)–2.949(4) | 2.612(4), 2.632(4) | N (1) 2.608(3), N(3) 2.436(3), O (1) 2.206(3) | 126.1(1) | 58.0(1) |
|
| 2.849(9) | 2.804(4)–2.880(4) | 2.583(4), 2.583(4) | N (1) 2.618(4), S (1) 2.766(1), S(2) 2.764(1) | 133.4(1) | 65.3(1) |
|
| 2.858(9) | 2.823(12)–2.907(12) | 2.600(12), 2.580(12) | N (1) 2.416(14), N(3) 2.633(7), S (1) 2.780(5) | 131.5(3) | 61.1(3) |
|
| Th (1) 2.861(9) | Th (1) 2.819(5)–2.899(6) | Th (1) 2.611(6), 2.579(6) | Th (1) N (1) 2.597(5), S (1) 2.982(1), S(1A) 3.039(1), S(2A) 2.889(1) | Th (1) 120.8(2) | Th (1) 59.9(1) |
|
| 2.874(18) | 2.813(9)–2.969(11) | 2.624(9), 2.598(9) | N (1) 2.674(10), N(3) 2.336(10), O (1) 2.271(8) | 128.2(3) | 76.7(3) |
|
| Th (1) 2.870(11) | Th (1) 2.824(7)–2.927(7) | Th (1) 2.619(7), 2.593(7) | Th (1) N (1) 2.562(6), O (1) 2.491(5), O(3A) 2.695(5), O(4A) 2.205(5) | Th (1) 124.0(2) | Th (1) 52.1(2) |
|
| 2.842(11) | 2.798(3)–2.886(3) | 2.581(3), 2.567(3) | N (1) 2.557(3), N(2) 2.565(3), O (1) 2.462(2), O(2) 2.452(2) | 136.3(1) | 52.4(1) |
|
| 2.859(7) | 2.827(9)–2.884(9) | 2.588(9), 2.595(9) | N (1) 2.427(11), N(2) 2.276(10), N(3) 2.650(5) | 133.8(2) | 59.1(3) |
|
| 2.850(13) | 2.786(5)–2.888(5) | 2.575(5), 2.592(5) | N (1) 2.503(4), N(2) 2.522(5), N(3) 2.512(4), C(35) 2.479(5) | 139.5(2) | 52.4(2) |
|
| 2.819(9) | 2.787(6)–2.867(6) | 2.559(6), 2.540(6) | N (1) 2.647(5), N(2) 2.463(5), N(4) 2.246(5) | 133.2(2) | 120.9(2) |
|
| 2.811(8) | 2.792(3)–2.851(3) | 2.554(3), 2.521(3) | N (1) 2.466(3), N(2) 2.440(3), N(3) 2.430(3) | 134.8(1) | 54.1(1) |
|
| 2.816(11) | 2.777(13)–2.865(16) | 2.541(13), 2.546(13) | N (1) 2.461(10), N(2) 2.468(11), N(3) 2.411(11) | 134.1(4) | 53.1(3) |
|
| 2.865(12) | 2.818(6)–2.927(6) | 2.611(6), 2.582(6) | N (1) 2.729(5), N(2) 2.513(4), N(3) 2.456(5), C(37) 2.496(6) | 128.8(2) | 51.4(2) |
|
| 2.820(5) | 2.796(5)–2.834(5) | 2.549(5), 2.552(5) | N (1) 2.334(4), N(2) 2.486(4), C(28) 2.467(5) | 135.5(2) | 32.2(1) |
|
| 2.823(7) | 2.791(4)–2.854(4) | 2.548(4), 2.557(4) | N (1) 2.356(4), N(2) 2.494(3), C(28) 2.480(4) | 132.4(1) | 30.1(1) |
|
| 2.834(7) | 2.801(6)–2.862(6) | 2.560(6), 2.568(6) | N (1) 2.368(4), N(2) 2.465(5), C(28) 2.443(5) | 133.3(2) | 30.7(2) |
|
| 2.879(17) | 2.811(15)–2.970(15) | 2.629(15), 2.599(15) | N (1) 2.350(16), N(2) 2.374(17), N(4) 2.651(8) | 131.1(4) | 56.9(5) |
|
| 2.832(6) | 2.811(5)–2.870(5) | 2.573(5), 2.554(5) | N (1) 2.344(4), N(2) 2.551(4), N(4) 2.496(4) | 134.0(1) | 51.8(1) |
|
| 2.841(7) | 2.809(5)–2.862(5) | 2.565(5), 2.583(5) | N (1) 2.304(5), N(2) 2.597(4), N(4) 2.494(4) | 138.3(2) | 52.1(1) |
|
| 2.825(8) | 2.798(3)–2.880(3) | 2.560(3), 2.548(3) | N (1) 2.318(3), N(4) 2.343(3) | 130.4(1) | 87.6(1) |
- —Deutsche Forschungsgemeinschaft10.13039/501100001659
- —National Natural Science Foundation of China10.13039/501100001809
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsOrganometallic Complex Synthesis and Catalysis · Radioactive element chemistry and processing · Radiopharmaceutical Chemistry and Applications
Introduction
Over the last four decades, terminal imido organoactinide complexes, with their characteristic AnN double bond, have captivated the scientific community due to their promise for advancing small molecule activation and/or catalysis. ?−? ? ? Although many of these complexes have been synthesized and their structures elucidated, fundamental questions remain concerning how structural nuances dictate reactivity. ?−? ? This inquiry not only drives the broader field of small molecule activation using organoactinide species? but also delves into the foundational aspects of actinide bonding, particularly the extent of covalency and the interplay between the 6d and 5f orbitals.? In our long-standing studies on thorium and uranium complexes that feature multiple bonds with actinides,? we have also reported terminal imido thorium complexes, including a notable example: the base-free terminal thorium imido metallocene, [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) (Figure S1), ?,? which readily activates a wide range of small molecules such as elemental sulfur and selenium, C–H bonds of pyridine derivatives, N–H bonds of amines, B–H bonds of boranes, Si–H bonds of silanes, Si–Cl bonds of chlorosilanes and last but not least the NN bond in diazoalkanes (Figure S1). ?,? Moreover, it plays a pivotal role as an intermediate in the catalytic hydroamination of internal acetylenes,? functions as an exceptionally efficient trimerization catalyst for PhCN,? and serves as a versatile precursor in the synthesis of terminal oxido and sulfido thorium metallocenes [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThE (E = O, S) by cycloaddition–elimination reactions with Ph_2_CE (E = O, S).? Motivated by the broad reactivity of these systems, we shifted our focus from the bulky 1,2,4-(Me_3_C)3_C_5_H_2 ligand to the widely used, less sterically demanding pentamethylcyclopentadienyl (C_5_Me_5) ligand. This transition allowed us to explore how reduced steric hindrance impacts the reactivity of thorium imido complexes, a decisive factor in organoactinide chemistry.? In this work, we present our findings on the synthesis, structure, and reactivity of the terminal thorium imido metallocene (η^5^-C_5_Me_5)_2_ThN(p-tolyl)(dmap)2 (5) and compare its behavior to that of its more sterically congested counterparts.?
Results and Discussion
Synthesis of (η5-C5Me5)2ThN(p-tolyl)(dmap)2 (5)
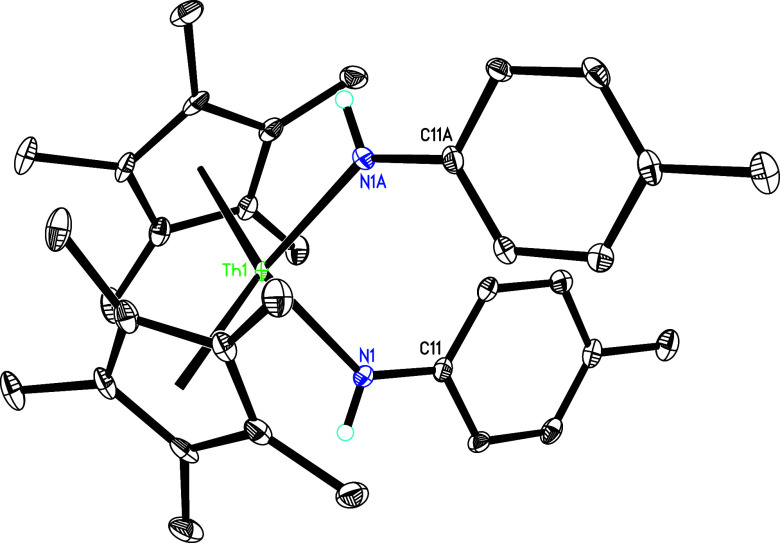
Treatment of the thorium dimethyl complex (η^5^-C_5_Me_5_)2_ThMe_2 (1) with 2 equiv of p-tolylNH_2_ affords the bis-amido complex (η^5^-C_5_Me_5_)2_Th(NH-p-tolyl)2 (2) quantitatively, as depicted in Scheme. The molecular structure of 2 is illustrated in Figure, with selected bond distances and angles listed in Table. Th(1)–N (1) distance is 2.329(3) Å, and the N(1)–Th(1)–N(1A) angle measures 103.5(2)°. Intriguingly, whereas prior studies demonstrated imido complex [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) formation from the reaction of dimethyl complex [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThMe_2 and bis-amido complex [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_Th(NH-p-tolyl)2, ?,? the treatment of (η^5^-C_5_Me_5)2_ThMe_2 (1) and (η^5^-C_5_Me_5)2_Th(NH-p-tolyl)2 (2) instead produces the μ-imido bridged bimetallic complex [(η^5^-C_5_Me_5)2_Th]2[μ-N(p-tolyl)]2 (3) (Scheme). This outcome reflects to lower steric hindrance of the C_5_Me_5 ligand compared to that of the 1,2,4-(Me_3_C)3_C_5_H_2 ligand. Moreover, complex 3 can also be synthesized upon reaction of (η^5^-C_5_Me_5)2_ThMe_2 (1) with 1 equiv of p-tolylNH_2_ (Scheme). The molecular structure of 3 is displayed in Figure, with selected bond distances and angles provided in Table. The Th(1)–N (1) and Th(1)–N(1A) distances are 2.271(5) and 2.401(5) Å, respectively, while the N(1)–Th(1)–N(1A) angle is 71.4(2)°. In a further reaction, treating (η^5^-C_5_Me_5_)2_ThMe_2 (1) with 1 equiv of p-tolylNH_2_ in the presence of pyridine results in the formation of the amido pyridyl complex (η^5^-C_5_Me_5_)2_Th[NH(p-tolyl)](κ^2^-C,N-C_5_H_4_N) (4) in quantitative conversion (Scheme). We propose that an initial imido pyridine adduct (η^5^-C_5_Me_5)2_ThN(p-tolyl)(py) is formed, which subsequently undergoes intramolecular deprotonation of the coordinated pyridine in α-position to furnish complex 4 (Scheme). The molecular structure of 4 is presented in Figure, and selected bond distances and angles are listed in Table. The Th(1)–N (1) and Th(1)–N(2) distances measure 2.355(5) and 2.475(6) Å, respectively, while the Th(1)–C(28) distance is 2.454(7) Å. This series of reactions underscores not only the versatile coordination chemistry of thorium complexes but also the delicate interplay of steric effects and reagent choice in directing the formation of distinct products. We then hypothesized that increasing the basicity of the Lewis base could help overcome these challenges and promote the formation of the desired terminal imido thorium metallocene. Indeed, the reaction of (η^5^-C_5_Me_5)2_ThMe_2 (1) with 1 equiv of p-tolylNH_2_ in the presence of 2 equiv of 4-dimethylaminopyridine (dmap) in toluene at room temperature forms the terminal imido complex (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5) in 86% isolated yield (Scheme). Moreover, contrary to (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap),? complex 5 crystallizes as an adduct with two equivalents of dmap, most likely reflecting the reduced steric hindrance of its p-tolyl group versus the mesityl analogue group. Nevertheless, when complex 1 is treated with 1 equiv of p-tolylNH_2 and 1 equiv of dmap, the reaction affords a complex mixture, and no pure compound can be isolated. This outcome further confirms our earlier conclusion that forming actinide imido metallocenes via methane elimination is highly sensitive to both the basicity of the coordinating Lewis base and the substituent effects on the cyclopentadienyl ring.? The molecular structure of 5 is shown in Figure, with selected bond distances and angles summarized in Table. The Th–N(2) and Th–N(4) distances, measured at 2.673(3) and 2.634(3) Å, respectively, are consistent with the presence of a dative nitrogen atom coordination. In stark contrast, the significantly shorter Th–N (1) bond of 2.082(3) Å mirrors the bond lengths typically observed in other thorium imido compounds such as [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) (2.038(3) Å),? (η^5^-C_5_Me_5)_2_ThN(mesityl)(dmap) (2.091(7) Å),? [η^5^-1,2,4-(Me_3_Si)3_C_5_H_2]_2_ThN(p-tolyl)(bipy) (2.073(6) Å) and [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) (2.090(7) Å). ?,? However, in contrast to the imido complex (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap),? while imido 5 is isolable as a solid, only the amido pyridyl complex (η^5^-C_5_Me_5)2_Th[NH(p-tolyl)][κ^2^-C,N-4-(Me_2_N)C_5_H_3_N] (5′) and dmap are observed by ^1^H NMR spectroscopy in C_7_D_8 solution over the temperature range of 20–100 °C. This observation suggests that the equilibrium lies toward the formation of 5′ and dmap, a phenomenon attributable to the reduced steric hindrance of the p-tolyl group compared with the mesityl analogue. Density functional theory (DFT) investigations suggest that the formation of 5′ begins with the transfer of an α-H atom from dmap to the imido ThN(p-tolyl) moiety via transition state TS5a, forming intermediate INT5 (Figure). In the following step, dmap dissociates from INT5 through the transition state TS5b, yielding product 5′. The conversion of 5 to 5′ + dmap is slightly exergonic (ΔG(298 K) = −1.6 kcal/mol) and encounters an overall barrier of ΔG ^‡^(298 K) = 21.6 kcal/mol. This energy profile suggests that an equilibrium between 5 and 5′ + dmap (Scheme) exists in solution and favors the side of 5′ + dmap, consistent with the NMR spectroscopic observations. This observation further implies that complex 5 is poised to exhibit a diverse reactivity pattern in small-molecule activation.
1: Selected Distances (Å) and Angles (°) for Compounds 2–23, 25–39, and 41–43
Molecular structure of 2 (thermal ellipsoids drawn at the 35% probability level).
Synthesis of Compounds 2–5
Molecular structure of 3 (thermal ellipsoids drawn at the 35% probability level).
Molecular structure of 4 (thermal ellipsoids drawn at the 35% probability level).
Molecular structure of 5 (thermal ellipsoids drawn at the 35% probability level).
Free energy profile (kcal/mol) for the reaction of 5 ⇌ 5′ + dmap. [Th] = (η5-C5Me5)2Th.
Synthesis of Compounds 6 and 7
Reactivity Studies
Next, we shift our focus to the reactivity of complex 5, juxtaposing the products from its reaction with various small molecules against those isolated from [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) (Figure S1), ?,? (η^5^-C_5_Me_5)_2_ThN(mesityl)(dmap) (Figure S2), ?,?,? [η^5^-1,3-(Me_3_C)2_C_5_H_3]_2_ThN(dipp)(dmap) and [η^5^-1,2,4-(Me_3_Si)3_C_5_H_2]_2_ThN(p-tolyl)(bipy) (Figures S3 and S4). ?,?,? To make comparisons easier for the reader, we have organized the reactions by substance class.
Reaction with Lewis Bases
In contrast to [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) (Figure S3), ?,? treatment of complex 5 or 5′ with 1-methylimidazole does not yield a simple 1-methylimidazole adduct. Instead, it produces the amido imidazolyl complex (η^5^-C_5_Me_5)2_Th[NH(p-tolyl)](κ^2^-C,N-1-MeC_3_H_2_N_2) (6) in quantitative conversion with concomitant dmap loss (Scheme), a result likely reflecting the reduced steric hindrance of the p-tolyl group compared with the 2,6-* ^i^ Pr_2_C_6_H_3_ group. The molecular structure of 6 is depicted in Figure, with selected bond distances and angles listed in Table. The Th–N (1) and Th–N(2) distances are 2.365(7) and 2.448(7) Å, respectively, whereas the Th–C(28) distance is 2.485(7) Å. Furthermore, proton transfer between complex 5 or 5′ and 2,6-Me_2_C_5_H_3_NO leads to the formation of the amido alkyl complex (η^5^-C_5_Me_5_)2_Th[NH(p-tolyl)](κ^2^-C,O-2-CH_2–6-MeC_5_H_3_NO) (7) (Scheme). The molecular structure of 7 is presented in Figure, while key bond distances and angles are summarized in Table. The Th–O (1) distance is 2.418(5) Å, whereas the Th–N(2) distance is 2.351(6) Å and Th–C(21) distance is 2.598(8) Å. Furthermore, unlike [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) (Figure S3), ?,? reacting complex 5 with Me_3_PO does not produce a Me_3_PO adduct. Instead, the reaction yields the bis-amido complex (η^5^-C_5_Me_5)2_Th[NH(p-tolyl)][κ^2^-C,N-N(p-tolyl)P(Me_2)CH_2_] (8) in 38% yield (Scheme), likely due to the reduced steric bulk imposed by the p-tolyl group compared to that of the 2,6- ^i^ *Pr_2_C_6_H_3_ group. We propose that complex 5 initially reacts with Me_3_PO to give a four-membered intermediate with the loss of dmap (Scheme). This intermediate then eliminates the oxido complex “(η^5^-C_5_Me_5_)_2_ThO” to generate the organic compound Me_3_PN(p-tolyl), which subsequently reacts with another molecule of 5 via proton transfer to form the product 8 along with dmap release. The molecular structure of 8 is shown in Figure, and selected bond distances and angles are listed in Table. The Th–N (1) and Th–N(2) distances are 2.489(6) and 2.348(5) Å, respectively, whereas the Th–C(28) is 2.700(7) Å.
Molecular structure of 6 (thermal ellipsoids drawn at the 35% probability level).
Molecular structure of 7 (thermal ellipsoids drawn at the 35% probability level).
Synthesis of Compound 8
Molecular structure of 8 (thermal ellipsoids drawn at the 35% probability level).
Reaction with Chalcogen Elements and Metal Halides
Moreover, like the thorium imido complex [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) (Figure S1), ?,? complex 5 reacts with 0.25 equiv of elemental sulfur (S_8) at 40 °C in toluene to form the four-membered complex (η^5^-C_5_Me_5_)2_Th[N(p-tolyl)SS](dmap) (9) with one dmap loss (Scheme). Figure shows the molecular structure of 9, while Table summarizes the selected bond distances and angles. The relatively long Th–N(2) distance of 2.661(4) Å is indicative of a datively coordinated nitrogen atom, while the Th–N (1) and Th–S (1) distances are 2.402(4) and 2.886 (1) Å, respectively. Nevertheless, in contrast to [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_Th[N(p-tolyl)SS] derived from [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) (Figure S1), ?,? complex 9 undergoes further reaction with 0.25 equiv of elemental sulfur (S_8) to form the six-membered complex (η^5^-C_5_Me_5)2_Th[N(p-tolyl)S_4] (10) with concomitant dmap release (Scheme), owing to the reduced steric bulk of the C_5_Me_5 ligand compared to that of the 1,2,4-(Me_3_C)3_C_5_H_2 ligand. Moreover, complex 10 can also be directly accessed via the reaction of 5 with 0.5 equiv of sulfur (S_8_) (Scheme). This reactivity contrasts with that of [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap), which forms the six-membered complex [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_Th[SSN(dipp)SS] (Figure S3), ?,? likely due to the diminished steric bulk of the p-tolyl substituent compared with the 2,6-* ^i^ *Pr_2_C_6_H_3 group. The molecular structure of 10 is shown in Figure, with selected bond distances and angles detailed in Table. The Th–N (1) and Th–S (1) distances are 2.380(11) and 2.819(4) Å, respectively, and the N(1)–Th-S (1) angle is 109.8(3)°. Nevertheless, in contrast to [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) forming a four-membered complex [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_Th[N(p-tolyl)SeSe] with selenium (Figure S1), ?,? when imido complex 5 is treated with 4 equiv of selenium in toluene at 40 °C, the six-membered complex (η^5^-C_5_Me_5)2_Th[N(p-tolyl)Se_4] (11) forms simultaneously with release of two equivalents of dmap (Scheme). This effect likely arises from the lower steric hindrance of the C_5_Me_5 ligand compared with the 1,2,4-(Me_3_C)3_C_5_H_2 ligand. Figure shows the molecular structure of 11, where Th–N (1) and Th–Se (1) distances are 2.351(8) and 2.927 (1) Å, respectively, and the N(1)–Th-Se (1) angle is 113.3(2)° (Table). However, treating 5 with selenium at 15 °C in toluene yields the amido selenido complex (η^5^-C_5_Me_5)2_Th[NH(p-tolyl)][κ^2^-N,Se-2-Se-4-(Me_2_N)C_5_H_3_N] (12) with one dmap loss (Scheme). In this case, selenium inserts into the pyridyl Th-[κ^2^-C,N-4-(Me_2_N)C_5_H_3_N] moiety, supporting the notion of an equilibrium between 5 and 5′ + dmap in solution. Figure displays the molecular structure of 12, and Table provides the selected bond distances and angles. The relatively long Th–N (1) distance of 2.590(4) Å is indicative of a datively coordinated nitrogen atom, while the Th–N(3) distance is 2.348(4) Å, and the Th–Se (1) is 2.959 (1) Å. Similarly, treatment of imido 5 with 1 equiv of tellurium in toluene at 120 °C produces the amido tellurido complex (η^5^-C_5_Me_5)_2_Th[NH(p-tolyl)][κ^2^-N,Te-2-Te-4-(Me_2_N)C_5_H_3_N] (13) with one dmap loss (Scheme), again indicating an equilibrium between 5 and 5′ + dmap in solution. Figure presents the molecular structure of 13, where the Th–N(2) distance of 2.594(5) Å signifies dative nitrogen atom coordination, the Th–N (1) distance is 2.333(5) Å, and the Th–Te (1) distance measures 3.162(2) Å (Table).
Synthesis of Compounds 9–13
Molecular structure of 9 (thermal ellipsoids drawn at the 35% probability level).
Molecular structure of 10 (thermal ellipsoids drawn at the 35% probability level).
Molecular structure of 11 (thermal ellipsoids drawn at the 35% probability level).
Molecular structure of 12 (thermal ellipsoids drawn at the 35% probability level).
Molecular structure of 13 (thermal ellipsoids drawn at the 35% probability level).
Like the imido compounds (η^5^-C_5_Me_5_)2_ThN(mesityl)(dmap) and [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) (Figures S2 and S3), ?,?,? complex 5 readily reacts with metal halides. For instance, treatment of 5 with 2 equiv of AgF yields the difluoride complex (η^5^-C_5_Me_5)2_ThF_2(dmap) (14) alongside other unidentified silver species that arise from the loss of nitrene (p-tolylN:) and one dmap molecule (Scheme). The molecular structure of 14 is shown in Figure, with key bond distances and angles summarized in Table. In particular, the Th–F (1) and Th–F(2) distances measure 2.182(5) and 2.184(5) Å, respectively, whereas the Th–N (1) bond extends to 2.607(6) Å, a distance that confirms the presence of a dative nitrogen atom coordination. Moreover, in contrast to compounds (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) and [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap), which, upon addition of copper halides, form heterobimetallic compounds (η^5^-C_5_Me_5)2_Th(X)[N(mesityl)Cu(dmap)] (X = Cl, Br) and [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_Th(Cl)[N(dipp)Cu(dmap)] (Figures S2 and S3), ?,?,? respectively, treatment of 5 with CuCl or CuBr results in the isolation of dichloride complex (η^5^-C_5_Me_5)2_ThCl_2(dmap)2 (15) and dibromide complex (η^5^-C_5_Me_5)2_ThBr_2(dmap) (16), respectively. This divergent reactivity is presumably due to the reduced steric hindrance imparted by the p-tolyl substituent compared with the mesityl and 2,6-* ^i^ *Pr_2_C_6_H_3 groups. The difluoride complex (η^5^-C_5_Me_5_)2_ThF_2(dmap) (14) and dibromide complex (η^5^-C_5_Me_5_)2_ThBr_2(dmap) (16) form adducts with one dmap molecule, but complex (η^5^-C_5_Me_5_)2_ThCl_2(dmap)2 (15) coordinates two dmap ligands. This difference likely arises from the varying steric protection around the metal atom offered by the coordinated ligands–in (η^5^-C_5_Me_5_)2_ThCl_2, there is sufficient space to accommodate two dmap ligands; by contrast, the shorter Th–F bond lengths or the bulkier bromide ligands impart greater steric shielding at the metal atom, precluding the coordination of two dmap ligands. The molecular structures of 15 and 16 are shown in Figures and ?, respectively, and selected bond distances and angles are provided in Table. In complex 15, the Th–Cl (1) and Th–Cl(2) distances are 2.732 (1) and 2.735 (1) Å, respectively, while the Th–N (1) and Th–N(3) distances are 2.746(4) and 2.737(4) Å, respectively, confirming a dative mode of nitrogen atom coordination. In complex 16, the Th–Br (1) and Th–Br(2) distances are 2.872 (1) and 2.921 (1) Å, respectively, whereas the Th–N (1) distance is 2.628(5) Å, further indicating that the nitrogen atom is coordinated in a dative fashion.
Molecular structure of 14 (thermal ellipsoids drawn at the 35% probability level).
Synthesis of Compounds 14–17
Molecular structure of 15 (thermal ellipsoids drawn at the 35% probability level).
Molecular structure of 16 (thermal ellipsoids drawn at the 35% probability level).
Reaction with Chlorosilanes and Silanes
Moreover, contrary to the thorium imido complex [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]_2_ThN(p-tolyl) and [η^5^-1,2,4-(Me_3_Si)3_C_5_H_2]_2_ThN(p-tolyl)(bipy) forming with PhSiH_2_Cl the amido chloride complexes [η^5^-1,2,4-(Me_3_E)3_C_5_H_2]2_Th(Cl)[N(p-tolyl)SiH_2_Ph] (E = C,? Si?) (Figures S1 and S4),? and (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) and [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) forming the dichloride complexes (η^5^-C_5_Me_5)2_ThCl_2(dmap) and [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThCl_2(dmap) (Figures S2 and S3), ?,?,? respectively, complex 5 reacts with PhSiH_2_Cl to give the chloro pyridyl complex (η^5^-C_5_Me_5)_2_Th(Cl)[κ^2^-C,N-4-(Me_2_N)C_5_H_3_N] (17) and p-tolylNHSiH_2_Ph in quantitative conversion with the loss of one dmap ligand (Scheme), in which the p-tolylNH moiety acts a nucleophile, attacking PhSiH_2_Cl. This reaction provides another compelling evidence for the established equilibrium between 5 and 5′ + dmap in the solution. The molecular structure of 17 is shown in Figure, and selected bond distances and angles are listed in Table. The Th–N (1) and Th–Cl (1) distances are 2.407(8) and 2.678(3) Å, respectively, whereas the Th–C(21) distance is 2.428(10) Å.
Molecular structure of 17 (thermal ellipsoids drawn at the 35% probability level).
Like the imido compounds [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) and (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) (Figures S1 and S2), ?,?,? complex 5 readily reacts with silanes. For example, treatment of complex 5 with 1 equiv of PhSiH_3 gives the amido phenyl complex (η^5^-C_5_Me_5_)2_Th[κ^3^-C,N,N-(4-Me_2_NC_5_H_3_N)SiH(Ph)N(4-MeC_6_H_3)] (18) with H_2_ and one dmap release (Scheme). Like the reaction observed for thorium imido complex [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) with PhSiH_3 (Figure S1), ?,? we propose that 5 initially reacts with PhSiH_3_ to give an amido hydride intermediate A while releasing dmap (Scheme; down). Next, this intermediate undergoes C–H activation of the p-tolyl group with concurrent H_2_ elimination, yielding an amido phenyl intermediate B that subsequently coordinates with the liberated dmap to form a dmap adduct C. Finally, this adduct experiences further C–H activation of the dmap ligand along with additional H_2_ release, ultimately delivering product 18 (Scheme). Alternatively, once the amido hydride intermediate A is generated, it effects C–H bond activation of the liberated dmap with concomitant H_2_ elimination to afford an amido-pyridyl intermediate D (Scheme; up). Intramolecular nucleophilic attack then delivers amido-hydride intermediate E, which undergoes a second C–H bond activation of the p-tolyl ring, with further H_2_ release, to furnish product 18 (Scheme). This reactivity is contrary to (η^5^-C_5_Me_5_)2_ThN(mesityl)(dmap) forming an amido alkyl dmap adduct (η^5^-C_5_Me_5)2_Th[κ^2^-N, C-{N(2-CH_2–4,6-Me_2_C_6_H_2_)(SiH_2_Ph)}](dmap) (Figure S2), ?,? attributed to the reduced steric hindrance imposed by the p-tolyl group compared with the mesityl group. Similarly, reacting imido complex 5 with 1 equiv of Ph_2_SiH_2_ yields the corresponding amido phenyl complex (η^5^-C_5_Me_5_)2_Th[κ^3^-C,N,N-(4-Me_2_NC_5_H_3_N)SiPh_2_N(4-MeC_6_H_3)] (19) with H_2_ and one dmap release (Scheme). The molecular structure of 18 is shown in Figure, whereas the molecular structure of 19 is provided in the Supporting Information. In complex 18, the Th–N (1), Th–N(2) and Th–C(21) distances are 2.356(4), 2.630(4) and 2.514(5) Å, respectively, which are comparable to those found in 19 (2.363(3), 2.624(3) and 2.488(4) Å, respectively).
Synthesis of Compounds 18 and 19
Molecular structure of 18 (thermal ellipsoids drawn at the 35% probability level).
Reaction with Alkynes
In analogy to [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) (Figure S1), ?,? complex 5 reacts with alkynes RCCR′. For example, treatment of complex 5 with PhCCPh gives a pyridyl amido complex (η^5^-C_5_Me_5)2_Th[N(p-tolyl)C(Ph)CHPh][κ^2^-C,N-4-(Me_2_N)C_5_H_3_N] (20) with the loss of one dmap ligand (Scheme). A plausible reaction mechanism begins with a [2 + 2] cycloaddition between 5 and PhCCPh. This is followed by deprotonation of an α-H on dmap, ultimately forming the final product 20 (Scheme). Nevertheless, imidos (η^5^-C_5_Me_5)_2_ThN(mesityl)(dmap) and [η^5^-1,3-(Me_3_C)2_C_5_H_3]_2_ThN(dipp)(dmap) do not show reactivity with PhCCPh, ?,? whereas complexes [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) (Figure S1) ?,? and 5 do. This effect is ascribed to the diminished steric bulk of the p-tolyl substituent relative to that of the mesityl and 2,6-* ^i^ *Pr_2_C_6_H_3 groups. The molecular structure of 20 is shown in Figure, and selected bond distances and angles are listed in Table. The Th–N (1) and Th–N(2) distances are 2.425(6) and 2.432(6) Å, respectively, whereas the Th–C(46) distance is 2.455(7) Å.
Synthesis of Compounds 20–22
Molecular structure of 20 (thermal ellipsoids drawn at the 35% probability level).
Reaction with Carbodiimides, Ketones, Isothiocyanates, CS2, Esters, and Amidates
In the presence of heterounsaturated organic substrates with polar C-E bonds, the thorium imido complexes [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) (Figure S1), ?,? (η^5^-C_5_Me_5)_2_ThN(mesityl)(dmap) (Figure S2), ?,? [η^5^-1,3-(Me_3_C)2_C_5_H_3]_2_ThN(dipp)(dmap) (Figure S3), ?,? [η^5^-1,2,4-(Me_3_Si)3_C_5_H_2]_2_ThN(p-tolyl)(bipy) (Figure S4), ?,? and 5 immediately react. In this case, although the specific reactivity patterns may vary, the overall reactivity is facilitated by the intrinsic steric properties of the thorium imido complexes and by the steric characteristics of the substrates employed. In analogy to imidos [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) and (η^5^-C_5_Me_5)_2_ThN(mesityl)(dmap) (Figures S1 and S2), ?,?,?
5 reacts with carbodiimides (RN)2_C to form the [2 + 2] cycloaddition products (η^5^-C_5_Me_5)2_Th[N(p-tolyl)C(NR)N(R)](dmap) (R = C_6_H_11 (21), * ^i^ *Pr (22)) besides one molecule of dmap (Scheme). This reactivity is contrary to [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) forming a four-membered metallaheterocycle [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_Th[N(C_6_H_11)C(Ndipp)N(C_6_H_11)] (Figure S3), ?,? in which [1,3]-Th migration occurs for the [2 + 2] addition intermediate,? a process driven by the greater steric hindrance of the * ^i^ *Pr_2_C_6_H_3_N moiety compared to mesityl and p-tolyl groups. The molecular structure of 21 is shown in Figure, whereas the molecular structure of 22 is provided in the Supporting Information. In complex 21, the Th–N (1), Th–N(3) and Th–N(5) distances are 2.658(4), 2.377(4) and 2.373(4) Å, respectively, which are comparable to those found in 22 (2.613(6), 2.365(5) and 2.344(6) Å, respectively).
Molecular structure of 21 (thermal ellipsoids drawn at the 35% probability level).
Moreover, complex 5 reacts with ketones. For example, when 5 is treated with 1 equiv of Ph_2_CO, it yields the [2 + 2] cycloaddition product (η^5^-C_5_Me_5_)_2_Th[N(p-tolyl)CPh_2_O](dmap) (23) with a concomitant loss of one dmap (Scheme). The disparate reactivity observed–where [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]_2_ThN(p-tolyl) forms a dmap supported terminal oxido product [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThO(dmap) in the presence of dmap (Figure S1), ?,? (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) leads to a four-membered metallaheterocycle (η^5^-C_5_Me_5)2_Th[OCPh_2_O](dmap) (Figure S2), ?,? complex 5 gives a [2 + 2] cycloaddition product (η^5^-C_5_Me_5)2_Th[N(p-tolyl)CPh_2_O](dmap) (23), and [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) yields a dimeric oxido complex {[η^5^-1,3-(Me_3_C)2_C_5_H_3]2_Th}2(μ-O)2 (Figure S3), ?,? –can be ascribed to differences in steric protection of the metal atom imposed by the coordinated ligands. In this context, complex 5 gives the [2 + 2] cycloaddition product (η^5^-C_5_Me_5)2_Th[N(p-tolyl)CPh_2_O](dmap) (23). This behavior reflects the lower steric bulk of the C_5_Me_5 ligand and p-tolyl substituent relative to the 1,2,4-(Me_3_C)3_C_5_H_2 ligand, mesityl, or 2,6-* ^i^ *Pr_2_C_6_H_3 groups. The molecular structure of 23, depicted in Figure and detailed in Table, features a relatively long Th–N (1) distance of 2.608(3) Å, indicative of a datively coordinated nitrogen atom. The Th–N(3) distance is 2.436(3) Å, whereas the Th–O (1) distance is 2.206(3) Å. Nevertheless, complex 23 is unstable and readily eliminates Ph_2_CN(p-tolyl) to form the monomeric terminal oxido (η^5^-C_5_Me_5)2_ThO(dmap). This intermediate further reacts with a second molecule of Ph_2_CO to produce the known four-membered metallaheterocycle (η^5^-C_5_Me_5)2_Th[OCPh_2_O](dmap) (24) (Scheme). Complex 24 can also be accessed directly through the reaction of 5 with 2 equiv of Ph_2_CO (Scheme). In a similar manner, a cycloaddition–elimination reaction occurs when 5 reacts with Ph_2_CS, furnishing the monomeric terminal sulfido (η^5^-C_5_Me_5)2_ThS(dmap), which immediately reacts with a second molecule of Ph_2_CS to yield the four-membered metallaheterocycle (η^5^-C_5_Me_5)_2_Th[SCPh_2_S](dmap) (25) (Scheme). The differences in reactivity between [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]_2_ThN(p-tolyl) (Figure S1), ?,? [η^5^-1,2,4-(Me_3_Si)3_C_5_H_2]_2_ThN(p-tolyl)(bipy) (Figure S4) ?,? and 5 forming the four-membered metallaheterocycle [η^5^-1,2,4-(Me_3_E)3_C_5_H_2]2_Th(SCPh_2_S) (E = C,? Si?) and (η^5^-C_5_Me_5)_2_Th[SCPh_2_S](dmap) (25), respectively, and [η^5^-1,3-(Me_3_C)2_C_5_H_3]_2_ThN(dipp)(dmap) yielding a dimeric sulfido complex {[η^5^-1,3-(Me_3_C)2_C_5_H_3]_2_Th}2(μ-S)2 (Figure S3), ?,? can also be attributed to the different steric protection of the metal atom exerted by the coordinated ligands, in which [η^5^-1,3-(Me_3_C)2_C_5_H_3]_2_Th fragment is not sufficiently sterically protected to prevent dimerization. The molecular structure of 25 is shown in Figure, and selected bond distances and angles are listed in Table. The Th–S (1) and Th–S(2) distances are 2.766 (1) and 2.764 (1) Å, whereas the Th–N (1) distance of 2.618(4) Å is indicative of a datively coordinated nitrogen atom.
Molecular structure of 23 (thermal ellipsoids drawn at the 35% probability level).
Synthesis of Compounds 23–25
Molecular structure of 25 (thermal ellipsoids drawn at the 35% probability level).
However, the four-membered metallaheterocycle (η^5^-C_5_Me_5_)2_Th[SCN(p-tolyl)NPh](dmap) (26) is isolated from the reaction of complex 5 and PhNCS, with one dmap being released (Scheme), where no cycloaddition–elimination reaction is observed. By analogy to the reactivity of (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) (Figure S2), ?,? formation of 26 may be explained by an initial [2 + 2] cycloaddition of 5 with PhNCS, followed by a [1,3]-Th migration (Scheme). The molecular structure of 26 is shown in Figure, with selected bond distances and angles provided in Table. The relatively long Th–N(3) distance of 2.633(7) Å is indicative of a datively coordinated nitrogen atom. The Th–N (1) and Th–S (1) distances measure 2.416(14) and 2.780(5) Å, respectively. In a related reaction, 5 reacts with CS_2 with concomitant dmap loss to initially form a [2 + 2] cycloaddition complex, which then dimerizes to yield [(η^5^-C_5_Me_5_)_2_Th]2{μ-[N(p-tolyl)C(S)S]}2 (27). The molecular structure of 27 is illustrated in Figure, and Table lists the selected bond distances and angles. The Th(1)–S (1), Th(1)–S(1A) and Th(1)–S(2A) distances are 2.982 (1), 3.039 (1) and 2.889 (1) Å, respectively, whereas the Th(1)–N (1) distance is 2.597(5) Å. Nevertheless, these reactivity patterns are in contrast to those observed for [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl), (η^5^-C_5_Me_5)_2_ThN(mesityl)(dmap) and [η^5^-1,3-(Me_3_C)2_C_5_H_3]_2_ThN(dipp)(dmap) (Figures S1–S3). ?,?,? Specifically, the thorium imido complex [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) reacts with CS_2 and PhNCS to form the [2 + 2] cycloaddition products [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_Th[N(p-tolyl)C(E)-S] (E = S, PhN) (Figure S1), ?,? while (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) affords the four-membered metallaheterocycle (η^5^-C_5_Me_5)2_Th[SCN(mesityl)-S](dmap) with CS_2 (Figure S2). ?,? In addition, [η^5^-1,3-(Me_3_C)2_C_5_H_3]_2_ThN(dipp)(dmap) gives rise to the dimer {[η^5^-1,3-(Me_3_C)2_C_5_H_3]2_Th}2(μ-S)2 via the reaction with both CS_2 and PhNCS (Figure S3). ?,? These differences can also be explained based on the steric arguments outlined above.
Molecular structure of 26 (thermal ellipsoids drawn at the 35% probability level).
Synthesis of Compounds 26 and 27
Molecular structure of 27 (thermal ellipsoids drawn at the 35% probability level).
Furthermore, when complex 5 is exposed to esters such as methyl methacrylate (MMA), a cycloaddition–elimination is not observed. Instead, as seen in the reactivity of [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) (Figure S1), ?,? the six-membered metallaheterocycle (η^5^-C_5_Me_5)2_Th[N(p-tolyl)CH_2_C(Me)C(OMe)O](dmap) (28) is furnished in quantitative conversion with the loss of one dmap molecule (Scheme). In this process, the ThN(p-tolyl) moiety serves as a nucleophile, undergoing a Michael addition reaction with MMA. This outcome contrasts with the formation of the methoxyl amidate complex [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_Th(OMe)[OC{C(Me)CH_2}N(dipp)] from [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) (Figure S3), ?,? due to the reduced steric bulk of the 1,3-(Me_3_C)2_C_5_H_3 ligand compared with 1,2,4-(Me_3_C)3_C_5_H_2 and C_5_Me_5 ligands. The molecular structure of 28 is shown in Figure, and selected bond distances and angles are summarized in Table. The relatively long Th–N (1) distance of 2.674(10) Å is indicative of a datively coordinated nitrogen atom. The Th–N(3) distance is 2.336(10) Å, whereas the Th–O (1) distance is 2.271(8) Å. Moreover, the reaction of 5 with rac-lactide does not yield a [2 + 2] cycloaddition product. Instead, the dimeric complex [(η^5^-C_5_Me_5)2_Th]2{μ-[OCH(Me)C(O)OCH(Me)C(N-p-tolyl)O]}2 (29) is formed along with dmap release (Scheme). This behavior contrasts with that of (η^5^-C_5_Me_5)_2_ThN(mesityl)(dmap) and [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) (Figures S2 and S3), ?,?,? which give the eight-membered metallaheterocycles (η^5^-C_5_Me_5)2_Th[OCH(Me)C(O)OCH(Me)C(Nmesityl)O] and [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_Th[OCH(Me)C(O)OCH(Me)C(Ndipp)O], respectively, again, presumably due to less steric hindrance imposed by the p-tolyl group compared with mesityl and 2,6-* ^i^ *Pr_2_C_6_H_3 groups. The molecular structure of 29 is presented in Figure, with selected bond distances and angles detailed in Table. The relatively long Th(1)–O(3A) distance of 2.695(5) Å is consistent with a datively coordinated oxygen atom, while the Th(1)–O (1) distance of 2.491(5) Å is longer than Th(1)–O(4A) (2.205(5) Å), likely a consequence of the steric repulsion between the p-tolyl group and C_5_Me_5 ligand. Moreover, the asymmetry in the Th(1)–O (1) (2.491(5) Å) vs Th(1)–N (1) (2.562(6) Å) bond distances suggests that the negative charge is predominantly localized on the O atom of the amidate fragment. Similarly, as observed in the reactivity of [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) (Figure S3), ?,? treatment of 5 with 2 equiv of the amidate PhCONH(p-tolyl) prompts deprotonation of the N–H moiety to yield the bis-amidate (η^5^-C_5_Me_5)2_Th[OC(Ph)N(p-tolyl)]2 (30) in quantitative conversion, with concomitant release of dmap and p-tolylNH_2 (Scheme). The molecular structure of 30 is shown in Figure, and selected bond distances and angles are summarized in Table. The Th–O bonds (Th–O (1) at 2.462(2) Å and Th–O(2) at 2.452(2) Å) are essentially identical, as are the Th–N bonds (Th–N (1) at 2.557(3) Å and Th–N(2) at 2.565(3) Å). Once again, the discrepancies in the Th–O and Th–N bond lengths reinforce the notion that the negative charge is mainly localized on the O atom of the amidate fragment.
Synthesis of Compounds 28–30
Molecular structure of 28 (thermal ellipsoids drawn at the 35% probability level).
Molecular structure of 29 (thermal ellipsoids drawn at the 35% probability level).
Molecular structure of 30 (thermal ellipsoids drawn at the 35% probability level).
Reaction with Organic Nitriles and Isonitriles
Moreover, in analogy to the imido complexes [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) (Figure S1), ?,? (η^5^-C_5_Me_5)_2_ThN(mesityl)(dmap) (Figure S2), ?,? [η^5^-1,3-(Me_3_C)2_C_5_H_3]_2_ThN(dipp)(dmap) (Figure S3), ?,? and [η^5^-1,2,4-(Me_3_Si)3_C_5_H_2]_2_ThN(p-tolyl)(bipy) (Figure S4), ?,? complex 5 also react with organic nitriles. For example, contrary to the reaction of PhCN with [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl), (η^5^-C_5_Me_5)_2_ThN(mesityl)(dmap) and [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) forming a [2 + 2] cycloaddition product [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_Th[N(p-tolyl)C(Ph)N], a dmap imido adduct (η^5^-C_5_Me_5)2_Th[NC(Ph)N(mesityl)](dmap) and an amido pyridyl complex [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_Th[NHC(Ph)Ndipp][κ^2^-C,N-4-(Me_2_N)C_5_H_3_N] (Figures S1–S3), ?,?,? respectively, reaction of 5 with 1 equiv of PhCN yields free dmap and crystals of both [2 + 2] cycloaddition product (η^5^-C_5_Me_5)2_Th[N(p-tolyl)C(Ph)N](dmap) (31a) and amidinyl pyridyl complex (η^5^-C_5_Me_5)2_Th[η^3^-NHC(Ph)N(p-tolyl)][κ^2^-C,N-4-(Me_2_N)C_5_H_3_N] (31b) (Scheme). These variations can also be attributed to different degrees of steric hindrance imposed on the thorium atom by the coordinated ligands, as previously described. Although both complexes 31a and 31b are isolable in the solid state, only the amidinyl pyridyl complex 31b is detected in the ^1^H NMR spectrum (recorded in C_7_D_8 solution between 20 and 100 °C). This observation suggests that the equilibrium is strongly shifted in favor of 31b. Moreover, DFT investigations indicate that the formation of 31b proceeds via an α-H transfer from dmap to the Th[N(p-tolyl)C(Ph)N] moiety through transition state TS31 (Figure). The conversion of 31b from 31a is slightly exergonic (ΔG(298 K) = −2.1 kcal/mol), but faces a reaction barrier of ΔG ^‡^(298 K) = 21.8 kcal/mol, which implies that if an equilibrium between 31a and 31b exists in the solution, it is ultimately shifted toward 31b–consistent with the NMR findings. The molecular structures of 31a and 31b are illustrated in Figures and ?, respectively, with selected bond distances and angles provided in Table. In complex 31a, the relatively long Th–N(3) distance of 2.650(5) Å is consistent with a datively coordinated nitrogen atom, whereas the Th–N (1) distance of 2.427(11) Å is longer than Th–N(2) (2.276(10) Å), likely attributed to the increased steric hindrance from the p-tolyl group and C_5_Me_5 ligand. In contrast, complex 31b exhibits that the Th–N (1), Th–N(2), and Th–N(3) distances are 2.503(4), 2.522(5), and 2.512(4) Å, respectively, with a Th–C(35) distance of 2.479(5) Å. Under similar reaction conditions, contrary to the reactivity of [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl),? treatment of 5 with 3 equiv of PhCN yields the eight-membered complex (η^5^-C_5_Me_5)2_Th[η^7^-N(p-tolyl)C(Ph)NC(Ph)NC(Ph)N] (32) with dmap elimination (Scheme), ostensibly due to the less steric hindrance of the C_5_Me_5 ligand compared with the 1,2,4-(Me_3_C)3_C_5_H_2 ligand. The molecular structure of 32 is shown in Figure, with selected bond distances and angles provided in Table. The Th–N (1) distance of 2.647(5) Å is longer than Th–N(2) (2.463(5) Å) and Th–N(4) (2.246(5) Å), again reflecting the steric interference between the p-tolyl group and C_5_Me_5 ligand.
Synthesis of Compounds 31 and 32
Free energy profile (kcal/mol) for the reaction of 31a ⇌ 31b. [Th] = (η5-C5Me5)2Th.
Molecular structure of 31a (thermal ellipsoids drawn at the 35% probability level).
Molecular structure of 31b (thermal ellipsoids drawn at the 35% probability level).
Molecular structure of 32 (thermal ellipsoids drawn at the 35% probability level).
Moreover, the thorium imido complexes [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) (Figure S3), ?,? [η^5^-1,2,4-(Me_3_Si)3_C_5_H_2]2_ThN(p-tolyl)(bipy) (Figure S4) ?,? and 5 exhibit identical reactivity toward benzyl nitrile PhCH_2_CN. In the case of 5, its reaction with PhCH_2_CN leads to the amidinyl iminato complex (η^5^-C_5_Me_5)2_Th[η^3^-N(p-tolyl)C(CH_2_Ph)NH](NCCHPh) (33) in quantitative conversion with concurrent dmap elimination (Scheme). We propose that complex 5 first undergoes a [2 + 2] cycloaddition with PhCH_2_CN, releasing dmap and generating a four-membered intermediate. This intermediate then reacts with a second molecule of PhCH_2_CN via deprotonation of the benzylic C–H bond, ultimately affording complex 33 (Scheme). A similar pathway occurs when 5 is exposed to Ph_2_CHCN, yielding the amidinyl iminato complex (η^5^-C_5_Me_5)2_Th[η^3^-N(p-tolyl)C(CHPh_2)NH](NCCPh_2) (34) in quantitative conversion with the loss of dmap (Scheme). This reactivity is contrary to [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) giving a mono-Cp amido iminato complex [η^5^-1,3-(Me_3_C)2_C_5_H_3]Th(NHdipp)(NCCPh_2)2(dmap)2 and [η^5^-1,2,4-(Me_3_Si)3_C_5_H_2]2_ThN(p-tolyl)(bipy) affording an μ-imido-bridged complex {[η^5^-1,2,4-(Me_3_Si)3_C_5_H_2]Th(NNCPh_2)(bipy)}2[μ-N(p-tolyl)]2 with Ph_2_CHCN (Figures S3 and S4), ?,?,? but similar to (η^5^-C_5_Me_5_)2_ThN(mesityl)(dmap) forming an iminato complex (η^5^-C_5_Me_5)2_Th[N(mesityl)C(CHPh_2)NH](NCCPh_2_) with Ph_2_CHCN (Figure S2), ?,? presumably due to the steric effect of the C_5_Me_5_ ligand. The molecular structure of 33 is shown in Figure, whereas the molecular structure of 34 is provided in the Supporting Information. The Th–N distances observed in 33 (2.466(3), 2.440(3) and 2.430(3) Å) are comparable to those found in 34 (2.461(10), 2.468(11) and 2.411(11) Å).
Synthesis of Compounds 33 and 34
Molecular structure of 33 (thermal ellipsoids drawn at the 35% probability level).
Moreover, analogously to the thorium imido complexes [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]_2_ThN(p-tolyl) (Figure S1), ?,? [η^5^-1,3-(Me_3_C)2_C_5_H_3]_2_ThN(dipp)(dmap) (Figure S3), ?,? and [η^5^-1,2,4-(Me_3_Si)3_C_5_H_2]_2_ThN(p-tolyl)(bipy) (Figure S4), ?,? complex 5 also reacts with organic isonitriles. However, unlike the reaction of 2,6-Me_2_C_6_H_3_NC with [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) forming an amido complex [η^5^-1,2,4-(Me_3_C)3_C_5_H_2][η^5^-1-(2,6-Me_2_C_6_H_3_NCCH_2_Me_2_C)-3,4-(Me_3_C)2_C_5_H_2]ThNH(p-tolyl) and with [η^5^-1,2,4-(Me_3_Si)3_C_5_H_2]2_ThN(p-tolyl)(bipy) affording an eight-membered heterocyclic compound [η^5^-1,2,4-(Me_3_Si)3_C_5_H_2]2_Th[N(p-tolyl)C(N-2,6-Me_2_C_6_H_3)C(H)N(6-MePh-2-CH_2)] (Figures S1 and S4), ?,?,? exposure of 5 toward 2,6-Me_2_C_6_H_3_NC in toluene at 40 °C yields the amido pyridyl complex (η^5^-C_5_Me_5)2_Th[N(p-tolyl)CHN(2,6-Me_2_C_6_H_3)][κ^2^-C,N-4-(Me_2_N)C_5_H_3_N] (35) with the elimination of one dmap ligand (Scheme). No intermediates were observed by NMR spectroscopy; however, drawing on the reactivity observed for the thorium phosphinidene complex [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThP(2,4,6-* ^t^ *Bu_3_Ph) with 2,6-Me_2_C_6_H_3_NC,? we propose that complex 5 initially undergoes a [2 + 1] cycloaddition with 2,6-Me_2_C_6_H_3_NC, with the concomitant loss of one dmap, to give a dmap metallaaziridine adduct (Scheme). Subsequent deprotonation of dmap yields complex 35. The molecular structure of 35 is shown in Figure, and selected bond distances and angles appear in Table. The relatively long Th–N (1) distance of 2.729(5) Å is indicative of a datively coordinated nitrogen atom. While the Th–N(2) and Th–N(3) distances are 2.513(5) and 2.456(5) Å, respectively, the Th–C(37) distance is 2.496(6) Å. In a separate reaction, treatment of complex 5 with 2,6-Me_2_C_6_H_3_NC at 15 °C in toluene affords an amido pyridyl complex (η^5^-C_5_Me_5)2_Th[NH(p-tolyl)][κ^2^-C,N-4-(Me_2_N)-6-(2,6-Me_2_C_6_H_3_NCH)C_5_H_2_N] (36) with one dmap lost (Scheme), further supporting an equilibrium between 5 and 5′ + dmap in solution. We propose that here, 2,6-Me_2_C_6_H_3_NC first inserts into the pyridyl Th-[κ^2^-C,N-4-(Me_2_N)C_5_H_3_N] moiety in 5′ to give an amido alkenyl intermediate. This intermediate subsequently undergoes deprotonation of the p-tolylNH group to yield an imido complex, which then converts via further deprotonation of the 4-(Me_2_N)-2-(2,6-Me_2_C_6_H_3_NCH)C_5_H_3_N moiety to furnish complex 36 (Scheme). The molecular structure of 36 is presented in Figure, with Table showing that the Th–N (1) and Th–N(2) distances are 2.334(4) and 2.486(4) Å, respectively, and the Th–C(28) bond measures 2.467(5) Å. Furthermore, insertion of isonitriles–specifically Me_3_CNC and C_6_H_11_NC–leads to the isolation of the amido alkenyl complexes (η^5^-C_5_Me_5)2_Th[NH(p-tolyl)][κ^2^-C,N-2-(Me_3_CNC)-4-(Me_2_N)C_5_H_3_N] (37) and (η^5^-C_5_Me_5)_2_Th[NH(p-tolyl)][κ^2^-C,N-2-(C_6_H_11_NC)-4-(Me_2_N)C_5_H_3_N] (38), respectively, each formed with the loss of one dmap (Scheme). This observation, once again, supports the presence of an equilibrium between 5 and 5′ + dmap in solution. The molecular structure of 37 is shown in Figure, whereas that of 38 is available in the Supporting Information. In complex 37, the Th–N (1), Th–N(2) and Th–C(28) distances are 2.356(4), 2.494(3) and 2.480(4) Å, respectively – values that are comparable to those found in 38 (2.368(4), 2.465(5) and 2.443(5) Å, respectively).
Molecular structure of 35 (thermal ellipsoids drawn at the 35% probability level).
Synthesis of Compounds 35 and 36
Molecular structure of 36 (thermal ellipsoids drawn at the 35% probability level).
Molecular structure of 37 (thermal ellipsoids drawn at the 35% probability level).
Synthesis of Compounds 37–39
Nevertheless, exposure of complex 5 to 2 equiv of PhCH_2_NC in toluene at 40 °C results in a four-membered heterocyclic complex (η^5^-C_5_Me_5_)_2_Th[N(p-tolyl)C(CHNCHPh)N(CH_2_Ph)](dmap) (39) with one dmap being released (Scheme). We suggest that 5 initially reacts with PhCH_2_NC via a [2 + 1] cycloaddition with one dmap loss to give a metallaaziridine, which rearranges to a four-membered heterometallacycle (Scheme). Within this intermediate, the reactive carbene fragment (RR′C:) subsequently couples with a second molecule of isonitrile PhCH_2_NC to afford the other four-membered heterometallacycle, which then undergoes a [1,3]-H migration to yield the complex 39 (Scheme). The molecular structure of 39 is provided in Figure, and selected bond distances and angles are detailed in Table. The Th–N (1) and Th–N(2) lengths are 2.350(16) and 2.374(17) Å, respectively, while the relatively long Th–N(4) distance of 2.651(8) Å is indicative of a datively coordinated nitrogen atom.
Molecular structure of 39 (thermal ellipsoids drawn at the 35% probability level).
Reaction with Organic Azides and Diazoalkane Derivatives
Moreover, analogously to the thorium imido complexes [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) (Figure S1), ?,? (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) (Figure S2), ?,? and [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) (Figure S3), ?,? compound 5 also reacts with organic azides. For example, contrary to [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap) affording a bis-amido complex [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_Th[NH(p-tolyl)][2-(dippN_3)-4-(Me_2_N)C_5_H_3_N] with p-tolylN_3 (Figure S3), ?,? but like [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) and (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) (Figures S1 and S2), ?,?,? exposure of complex 5 toward p-tolylN_3 in toluene at 40 °C eliminates dmap and gives the known tetraazametallacyclopentene (η^5^-C_5_Me_5)2_Th[N(p-tolyl)NNN(p-tolyl)] (40) (Scheme), presumably due to the less steric hindrance introduced by the p-tolyl group compared with the 2,6-* ^i^ *Pr_2_C_6_H_3 group. However, when this reaction is carried out in toluene at 15 °C, dmap is released and the bis-amido complex (η^5^-C_5_Me_5)2_Th[NH(p-tolyl)][κ^2^-N,N-2-N(NN-p-tolyl)-4-(Me_2_N)C_5_H_3_N] (41) is isolated (Scheme), once again, providing support for the equilibrium between 5 and 5′ + dmap in the solution. A similar reactivity is observed when Ph_3_CN_3 is added to complex 5, resulting in the bis-amido complex (η^5^-C_5_Me_5_)2_Th[NH(p-tolyl)][κ^2^-N,N-2-N(NNCPh_3)-4-(Me_2_N)C_5_H_3_N] (42) in quantitative conversion with the loss of one molecule of dmap (Scheme). The molecular structure of 41 is shown in Figure, while the molecular structure of 42 is provided in the Supporting Information. In complex 41, the Th–N (1), Th–N(2) and Th–N(4) distances measure 2.344(4), 2.551(4) and 2.496(4) Å, respectively–values that are comparable to those found in 42 (2.304(5), 2.597(4) and 2.494(4) Å, respectively). In contrast, treatment of complex 5 with Me_3_SiN_3_ yields the azido amido complex (η^5^-C_5_Me_5_)2_Th(N_3)[N(p-tolyl)SiMe_3_] (43) along with dmap in quantitative conversion (Scheme), wherein the ThN(p-tolyl) moiety serves as a nucleophile. This reactivity diverges from that observed with [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_ThN(dipp)(dmap), which forms a bis-azido complex [η^5^-1,3-(Me_3_C)2_C_5_H_3]2_Th(N_3)2(dmap)2 (Figure S3), ?,? presumably due to the increased steric hindrance imparted by the C_5_Me_5 ligand compared with the 1,3-(Me_3_C)2_C_5_H_3 ligand. The molecular structure of 43 is provided in Figure, with selected bond distances and angles reported in Table. The Th–N (1) distance is 2.318(3) Å, whereas Th–N(4) distance is 2.343(3) Å. Moreover, the N(1)–Th-N(4) angle is 87.6 (1)°.
Synthesis of Compounds 40–43
Molecular structure of 41 (thermal ellipsoids drawn at the 35% probability level).
Molecular structure of 43 (thermal ellipsoids drawn at the 35% probability level).
Moreover, analogous to the reactivity observed for the thorium imido complexes [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) (Figure S1), ?,? and (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) (Figure S2), ?,? complex 5 also engages with diazoalkanes. However, contrary to [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) forming the amido nitrilimido complex [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_Th[NH(p-tolyl)](N_2_CSiMe_3) with Me_3_SiCHN_2 (Figure S1), ?,? but similar to (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) (Figure S2), ?,? no amido nitrilimido complex is produced. Instead, the known bimetallic compound [(η^5^-C_5_Me_5)2_Th]2(μ-NNNCSiMe_3)2 (44) is isolated (Scheme), a result that is presumably attributed to the reduced steric bulk of the C_5_Me_5_ ligand compared to the 1,2,4-(Me_3_C)3_C_5_H_2 ligand. We propose the following mechanism: Complex 5 initially reacts with Me_3_SiCHN_2_ to generate a three-membered complex when liberating dmap. This intermediate undergoes a [1,3]-C migration to yield p-tolyl complex, which immediately experiences either inter- or intramolecular deprotonation of an α-H from the Me_3_SiCHN_3_ group to yield complex 44 and toluene as a byproduct (Scheme).
Synthesis of Compound 44
Conclusions
In summary, we have synthesized and fully characterized the Lewis base supported thorium imido metallocene, (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5). In toluene solution, an equilibrium is established between 5, dmap and the amido pyridyl complex (η^5^-C_5_Me_5)2_Th[NH(p-tolyl)][κ^2^-C,N-4-(Me_2_N)C_5_H_3_N] (5′), highlighting the complex’s inherent versatility. Complex 5 engages a broad spectrum of substrates–ranging from 1-methylimidazole, pyridine-N-oxide derivative 2,6-Me_2_C_5_H_3_NO, Me_3_PO, elemental sulfur (S_8) and selenium (Se), metal halides, silanes, and alkynes, to carbodiimides, ketones, thio-ketones, isothiocyanates, CS_2_, esters, amidates, organic nitriles and isonitriles, and organic azides, thus affording a diverse array of products. These include amido imidazolyl complex, amido alkyl complex, and bis-amido complex, as well as heterometallacycles of various ring sizes (four-, five-, six- or eight-membered), dihalido species, bis-amidate compounds, amidinyl iminato complexes, amido phenyl, amido pyridyl, and amido alkenyl derivatives. Moreover, reactions with elemental selenium (Se) and tellurium (Te), chlorosilane PhSiH_2_Cl, organic isonitriles (2,6-Me_2_C_6_H_3_NC, Me_3_CNC and C_6_H_11_NC), and organic azides (p-tolylN_3_ and Ph_3_CN_3_) further underscore the remarkable potential of complex 5′ in small molecule activation forming the amido selenido compound (η^5^-C_5_Me_5_)2_Th[NH(p-tolyl)][κ^2^-N,Se-2-Se-4-(Me_2_N)C_5_H_3_N] (12), amido tellurido species (η^5^-C_5_Me_5)2_Th[NH(p-tolyl)][κ^2^-N,Te-2-Te-4-(Me_2_N)C_5_H_3_N] (13), chloro pyridyl complex (η^5^-C_5_Me_5)2_Th(Cl)[κ^2^-C,N-4-(Me_2_N)C_5_H_3_N] (17), amido pyridyl complex (η^5^-C_5_Me_5)2_Th[NH(p-tolyl)][κ^2^-C,N-4-(Me_2_N)-6-(2,6-Me_2_C_6_H_3_NCH)C_5_H_2_N] (36), amido alkenyl complexes (η^5^-C_5_Me_5)2_Th[NH(p-tolyl)][κ^2^-C,N-2-(Me_3_CNC)-4-(Me_2_N)C_5_H_3_N] (37) and (η^5^-C_5_Me_5)2_Th[NH(p-tolyl)][κ^2^-C,N-2-(C_6_H_11_NC)-4-(Me_2_N)C_5_H_3_N] (38), and bis-amido complexes (η^5^-C_5_Me_5)2_Th[NH(p-tolyl)][κ^2^-N,N-2-N(NN-p-tolyl)-4-(Me_2_N)C_5_H_3_N] (41) and (η^5^-C_5_Me_5)2_Th[NH(p-tolyl)][κ^2^-N,N-2-N(NNCPh_3)-4-(Me_2_N)C_5_H_3_N] (42), respectively. Furthermore, while mixing 5 with Me_3_SiN_3_ affords the azido amido complex (η^5^-C_5_Me_5_)2_Th(N_3)[N(p-tolyl)SiMe_3_] (43), the bimetallic complex [(η^5^-C_5_Me_5_)2_Th]2(μ-NNNCSiMe_3)2 (44) and toluene are formed when 5 is exposed to Me_3_SiCHN_2_.
Moreover, while these thorium imido metallocenes share similar overall reactivity patterns, subtle variations in the cyclopentadienyl ligand can fine-tune their individual reactivity patterns. For example, imido [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) is stable, ?,? whereas (η^5^-C_5_Me_5)_2_ThN(p-tolyl) is not. While the reaction of [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) with S_8 or Se gives four-membered metallaheterocycles [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_Th[N(p-tolyl)EE] (E = S, Se) (Figure S1), ?,?,? compound 5 yields the six-membered complex (η^5^-C_5_Me_5)2_Th[N(p-tolyl)E_4] (E = S (10), Se (11)). The amido hydrido complex [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_Th(H)[N(p-tolyl)SiH_2_Ph] formed from [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) and PhSiH_3 is stable (Figure S1), ?,? whereas a C–H bond activation of the p-tolyl ligand occurs for the product derived from 5 and PhSiH_3. Moreover, while the [2
- 2] cycloaddition product (η^5^-C_5_Me_5_)_2_Th[N(p-tolyl)CPh_2_O](dmap) (23) can be isolated from the reaction of complex 5 with 1 equiv of Ph_2_CO, this is not possible for [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]_2_ThN(p-tolyl).? Instead, the terminal oxido dmap adduct [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]_2_ThO(dmap) is generated in the reaction between [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]_2_ThN(p-tolyl) and Ph_2_CO in the presence of dmap (Figure S1). ?,? Furthermore, treatment of [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]_2_ThN(p-tolyl) with PhNCS affords a formal [2 + 2]-cycloaddition product [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_Th[N(p-tolyl)C(NPh)-S] (Figure S1), ?,? whereas (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) (Figure S2) ?,? and compound 5 yield the four-membered metallaheterocycles (η^5^-C_5_Me_5)2_Th[SCN(mesityl)NPh](dmap) and (η^5^-C_5_Me_5)_2_Th[SCN(p-tolyl)NPh](dmap) (26), respectively. While exposure of [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) toward CS_2 furnishes the formal [2 + 2]-cycloaddition product [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_Th[N(p-tolyl)C(S)-S] (Figure S1), ?,? the imido complexes (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) (Figure S2) ?,? and 5 give either a four-membered metallaheterocycle (η^5^-C_5_Me_5)2_Th[SCN(mesityl)-S](dmap) or a dimeric complex [(η^5^-C_5_Me_5)_2_Th]2{μ-[N(p-tolyl)C(S)S]}2 (27). The imido compound [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) is an effective catalyst for the trimerization of PhCN,? whereas 5 reacts with 3 equiv of PhCN to afford an eight-membered complex (η^5^-C_5_Me_5)2_Th[η^7^-N(p-tolyl)C(Ph)NC(Ph)NC(Ph)N] (32). Furthermore, the amido complex [η^5^-1,2,4-(Me_3_C)3_C_5_H_2][η^5^-1-(2,6-Me_2_C_6_H_3_NCCH_2_Me_2_C)-3,4-(Me_3_C)2_C_5_H_2]ThNH(p-tolyl) is isolated from the reaction of [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) with 2,6-Me_2_C_6_H_3_NC (Figure S1), ?,? but 5 produces an amido pyridyl complex (η^5^-C_5_Me_5)2_Th[N(p-tolyl)CHN(2,6-Me_2_C_6_H_3)][κ^2^-C,N-4-(Me_2_N)C_5_H_3_N] (35). Although the reaction of [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) with Me_3_SiCHN_2 results in the formation of an amido nitrilimido complex [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_Th[NH(p-tolyl)](N_2_CSiMe_3) (Figure S1), ?,? complexes (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) (Figure S2) ?,? and 5 give a bimetallic complex [(η^5^-C_5_Me_5)2_Th]2(μ-NNNCSiMe_3)2 (44) concomitant with the elimination of mesitylene and toluene, respectively.
However, the substituents on the imido group also influence the reactivity of these Th imido compounds. For example, (η^5^-C_5_Me_5_)2_ThN(mesityl)(dmap) forms as a mono-dmap adduct,? whereas complex 5 crystallizes as an adduct with two coordinated dmap ligands. While in the C_7_D_8 solution, an equilibrium between imido 5 and amido pyridyl complex 5′ cannot be detected by ^1^H NMR spectroscopy, it is observable for (η^5^-C_5_Me_5_)2_ThN(mesityl)(dmap).? Reaction of (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) with CuCl or CuBr produce the heterobimetallic compounds (η^5^-C_5_Me_5)2_Th(X)[N(mesityl)Cu(dmap)] (X = Cl, Br) (Figure S2), ?,? but 5 yields the dichloride complex (η^5^-C_5_Me_5)2_ThCl_2(dmap)2 (15) and the dibromide complex (η^5^-C_5_Me_5_)2_ThBr_2(dmap) (16), respectively. Moreover, while reaction of (η^5^-C_5_Me_5_)2_ThN(mesityl)(dmap) with PhSiH_3 forms an amido alkyl dmap adduct (η^5^-C_5_Me_5_)2_Th[κ^2^-N,C-{N(2-CH_2–4,6-Me_2_C_6_H_2_)(SiH_2_Ph)}](dmap) (Figure S2), ?,? complex 5 gives an amido phenyl complex (η^5^-C_5_Me_5_)2_Th[κ^3^-C,N,N-(4-Me_2_NC_5_H_3_N)SiH(Ph)N(4-MeC_6_H_3)] (18). Compounds [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) (Figure S1) ?,? and 5 immediately react with PhCCPh, but (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) does not.? In addition, while the [2 + 2] cycloaddition product (η^5^-C_5_Me_5)2_Th[N(p-tolyl)CPh_2_O](dmap) (23) is isolated from the reaction of complex 5 with 1 equiv of Ph_2_CO, this is not possible for (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) (Figure S2). ?,? Exposure of (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) toward CS_2 results in a four-membered metallaheterocycle (η^5^-C_5_Me_5_)2_Th[SCN(mesityl)-S](dmap) (Figure S2) ?,? but complex 5 affords the dimeric complex [(η^5^-C_5_Me_5)2_Th]2{μ-[N(p-tolyl)C(S)S]}2 (27). Moreover, while reaction of (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) with rac-lactide gives an eight-membered metallaheterocycle (η^5^-C_5_Me_5)2_Th[OCH(Me)C(O)OCH(Me)C(Nmesityl)O] (Figure S2), ?,? complex 5 yields a dimeric product [(η^5^-C_5_Me_5)2_Th]2{μ-[OCH(Me)C(O)OCH(Me)C(N-p-tolyl)O]}2 (29). Reaction of (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) with PhCN affords a dmap imido adduct (η^5^-C_5_Me_5)_2_Th[NC(Ph)N(mesityl)](dmap) (Figure S2), ?,? whereas [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]_2_ThN(p-tolyl) forms a [2 + 2] cycloaddition product [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_Th[N(p-tolyl)C(Ph)N] (Figure S1), ?,? and complex 5 gives a mixture of a [2 + 2] cycloaddition product (η^5^-C_5_Me_5)2_Th[N(p-tolyl)C(Ph)N](dmap) (31a) and amidinyl pyridyl complex (η^5^-C_5_Me_5)2_Th[η^3^-NHC(Ph)N(p-tolyl)][κ^2^-C,N-4-(Me_2_N)C_5_H_3_N] (31b). Although the tetraazametallacyclopentenes [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_Th[N(p-tolyl)NNN(p-tolyl)] (Figure S1) ?,? and (η^5^-C_5_Me_5)2_Th[N(p-tolyl)NNN(p-tolyl)] (40) are isolated as stable products from the reactions of [η^5^-1,2,4-(Me_3_C)3_C_5_H_2]2_ThN(p-tolyl) and complex 5 with p-tolylN_3, respectively, NN cleavage and mesitylN_3 elimination are encountered in the reaction of (η^5^-C_5_Me_5)2_ThN(mesityl)(dmap) with p-tolylN_3 (Figure S2). ?,? Further exploration of actinide imido complexes is ongoing, and the results will be reported in due course.
Experimental Section
General Procedures
All reactions and product manipulations were conducted under an atmosphere of dry dinitrogen using standard Schlenk or cannula techniques or in a glovebox, ensuring rigid exclusion of air and moisture. Organic solvents were freshly distilled from sodium benzophenone ketyl immediately before use. (η^5^-C_5_Me_5_)2_ThMe_2 (1) ?,? was prepared according to the literature method, and all other chemicals were purchased from Aldrich Chemical Co. and Beijing Chemical Co. and used as received unless stated otherwise. Infrared spectra were recorded in KBr pellets on an Avatar 360 Fourier transform spectrometer. ^1^H and ^13^C{^1^H} NMR spectra were recorded on a Bruker AV 400 (at 400 and 100 MHz, respectively), or a JEOL AV 400 (at 400 and 100 MHz, respectively), or a JEOL 600 (at 600 and 151 MHz, respectively). Chemical shifts are reported in δ units and referenced to the residual protons of the deuterated solvents, which served as internal standards, for proton and carbon chemical shifts. ^31^P{^1^H} NMR spectra were recorded on a JEOL AV 400 at 162 MHz. ^29^Si{^1^H}, ^77^Se{^1^H}, and ^125^Te{^1^H} NMR spectra were recorded on a JEOL 600 at 119.2, 114, and 189 MHz, respectively. Phosphorus, silicon, selenium, and tellurium chemical shifts were referenced to external 85% H_3_PO_4_ (0.00 ppm), Me_4_Si (0.00 ppm), Me_2_Se (0.00 ppm), and Me_2_Te (0.00 ppm), respectively. Melting points were determined on an X-6 melting point apparatus and were uncorrected. Elemental analyses were performed on a Vario EL elemental analyzer.
Caution: Natural thorium (primary isotope ^232^Th) is a weak α-emitter (4.012 MeV) with a half-life of 1.41 × 10^10^ years. Therefore, manipulations and reactions should be carried out in monitored fume hoods or in an inert-atmosphere drybox in a laboratory equipped with α- and β-counting equipment. Moreover, all organic reactants used in this work are standard or commercially available reagents, and there is no special safety consideration.
Preparation of (η5-C5Me5)2Th(NH-p-tolyl)2 (2)
Method A
A toluene (10 mL) solution of p-toluidine (0.43 g, 4.0 mmol) was added to a stirred toluene (20 mL) solution of (η^5^-C_5_Me_5_)2_ThMe_2 (1; 1.06 g, 2.0 mmol) at room temperature. The resulting mixture was stirred overnight at room temperature. Followed by removal of the solvent, the residue was extracted with benzene (3 × 10 mL) and filtered. The volume of the combined filtrates was reduced to 10 mL, and colorless crystals of 2 developed when this solution was kept at 10 °C for 1 day. The crystals of 2 were then collected by filtration, rapidly washed with cold n-hexane (5 mL), and dried under vacuum at room temperature overnight. Yield: 1.34 g (94%). M.p.: 188–190 °C (dec.). ^1^H NMR (C_6_D_6_): δ 6.92 (d, J = 6.8 Hz, 4H, phenyl), 6.71 (d, J = 7.0 Hz, 4H, phenyl), 5.17 (s, 2H, NH), 2.20 (s, 6H, tolylCH 3), 1.95 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 151.6 (phenyl C), 129.9 (phenyl C), 126.0 (phenyl C), 124.8 (phenyl C), 118.0 (ring C), 20.7 (tolylCH_3_), 11.3 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 3315 (m), 2965 (s), 2917 (s), 1605 (s), 1503 (s), 1404 (s), 1383 (s), 1262 (s), 1108 (s), 1021 (s), 812 (s). Anal. Calcd for C_34_H_46_N_2_Th: C, 57.13; H, 6.49; N, 3.92. Found: C, 57.16; H, 6.52; N, 3.90.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of p-toluidine (4.3 mg, 0.04 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThMe_2 (1; 10.6 mg, 0.02 mmol) and C_6_D_6_ (0.2 mL). Resonances of 2 and that of methane were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of [(η5-C5Me5)2Th]2[μ-N(p-tolyl)]2·C6H6 (3·C6H6)
Method A
A toluene (10 mL) solution of (η^5^-C_5_Me_5_)2_Th(NH-p-tolyl)2 (2; 0.72 g, 1.0 mmol) was added to a stirred toluene (20 mL) solution of (η^5^-C_5_Me_5)2_ThMe_2 (1; 0.53 g, 1.0 mmol) at room temperature. The mixture was then stirred at 80 °C overnight and subsequently cooled to room temperature. After removal of the solvent under reduced pressure, the resulting residue was extracted with benzene (3 × 10 mL) and filtered. The combined filtrate was concentrated to 10 mL, and yellow crystals of 3·C_6_H_6_ formed when this solution was maintained at 10 °C for 1 day. Crystals of 3·C_6_H_6_ were isolated by filtration, rapidly washed with cold n-hexane (5 mL), and dried under vacuum at room temperature overnight. Yield: 1.14 g (88%). M.p.: 178–180 °C (dec.). ^1^H NMR (C_6_D_6_): δ 7.19 (d, J = 7.4 Hz, 4H, phenyl), 7.15 (s, 6H, C_6_ H 6), 6.54 (d, J = 7.4 Hz, 4H, phenyl), 2.36 (s, 6H, tolylCH 3), 2.10 (s, 60H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 150.0 (phenyl C), 131.9 (phenyl C), 128.5 (C 6_H_6), 127.2 (phenyl C), 125.9 (phenyl C), 119.4 (ring C), 20.7 (tolylCH_3_), 13.3 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2961 (s), 2906 (s), 2856 (s), 1598 (m), 1435 (s), 1384 (s), 1259 (s), 1089 (s), 1019 (s), 804 (s). Anal. Calcd for C_60_H_80_N_2_Th_2_: C, 55.72; H, 6.23; N, 2.17. Found: C, 55.74; H, 6.21; N, 2.15.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solutionof p-toluidine (2.1 mg, 0.02 mmol) was slowly addedto a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThMe_2 (1; 10.6mg, 0.02 mmol) and C_6_D_6_ (0.2 mL). Resonancesof 3 and that of methane were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was keptat 80 °C overnight.
Preparation of (η5-C5Me5)2Th[NH(p-tolyl)](κ2-C,N-C5H4N) (4)
Method A
A toluene (10 mL) solution of p-toluidine (0.21 g, 2.0 mmol) and pyridine (0.16 g, 2.0 mmol) was added to a stirred toluene (20 mL) solution of (η^5^-C_5_Me_5_)2_ThMe_2 (1; 1.06 g, 2.0 mmol) at room temperature. The reaction mixture was then stirred at 80 °C overnight. After cooling, the solvent was removed under reduced pressure, and the resulting residue was extracted with benzene (3 × 10 mL) and filtered. The volume of the combined filtrate was reduced to 10 mL, and colorless crystals of 4 formed when this solution was kept at 10 °C for 1 day. These crystals were isolated by filtration, rapidly washed with cold n-hexane (5 mL), and subsequently dried under vacuum at room temperature overnight. Yield: 1.24 g (90%). M.p.: 130–132 °C (dec.). ^1^H NMR (C_6_D_6_): δ 8.27 (d, J = 5.2 Hz, 1H, py), 7.78 (d, J = 7.3 Hz, 1H, py), 7.13 (m, 3H, py and phenyl), 6.81 (d, J = 8.4 Hz, 2H, phenyl), 6.59 (m, 1H, py), 4.62 (s, 1H, NH), 2.38 (s, 3H, tolylCH 3), 1.89 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 238.2 (ThC), 155.2 (aryl C), 145.0 (aryl C), 136.1 (aryl C), 131.0 (aryl C), 129.6 (aryl C), 124.0 (aryl C), 123.3 (aryl C), 122.8 (aryl C), 118.0 (ring C), 20.8 (tolylCH_3_), 11.2 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2921 (s), 2856 (s), 1585 (s), 1440 (s), 1384 (s), 1260 (s), 1092 (s), 1020 (s), 800 (s). Anal. Calcd for C_32_H_42_N_2_Th: C, 55.97; H, 6.16; N, 4.08. Found: C, 55.96; H, 6.14; N, 4.11.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of p-toluidine (2.1 mg, 0.02 mmol) and pyridine (1.6 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThMe_2 (1; 10.6 mg, 0.02 mmol) and C_6_D_6_ (0.2 mL). Resonances of 4 and that of methane were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 80 °C overnight.
Preparation of (η5-C5Me5)2ThN(p-tolyl)(dmap)2 (5)
A toluene (10 mL) solution of p-toluidine (0.21 g, 2.0 mmol) and dmap (0.50 g, 4.1 mmol) was added to a stirred toluene (20 mL) solution of (η^5^-C_5_Me_5_)2_ThMe_2 (1; 1.06 g, 2.0 mmol) at room temperature. The reaction mixture was stirred overnight at ambient temperature. After removal of the solvent under reduced pressure, the residue was extracted with benzene (3 × 10 mL) and filtered. The filtrate was concentrated to 10 mL, and yellow crystals of complex 5 formed upon storage at 10 °C for 1 day. The crystals were then isolated by filtration, rapidly washed with 5 mL of cold n-hexane, and dried under vacuum at room temperature overnight. Yield: 1.47 g (86%). M.p.: 138–140 °C (dec.). ^1^H NMR (C_6_D_6_): δ 8.46 (d, J = 3.8 Hz, 2H, dmap), 8.15 (d, J = 6.0 Hz, 1H, py), 7.17 (m, 3H, phenyl and py), 6.91 (d, J = 8.0 Hz, 2H, phenyl), 6.09 (d, J = 5.4 Hz, 2H, dmap), 6.05 (m, 1H, py), 4.62 (s, 1H, NH), 2.40 (s, 3H, tolylCH 3), 2.34 (s, 6H, N(CH 3)2), 2.21 (s, 6H, (CH 3)2_N, dmap), 2.01 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6): δ 234.2 (ThC), 155.9 (aryl C), 154.8 (aryl C), 154.3 (dmap C), 150.4 (dmap C), 143.9 (aryl C), 129.6 (aryl C), 122.8 (aryl C), 122.4 (ring C), 118.2 (aryl C), 110.9 (aryl C), 108.7 (aryl C), 106.7 (dmap C), 38.6 (NCH_3_, dmap), 38.2 (NCH_3_), 20.9 (tolylCH_3_), 11.3 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2907 (s), 1608 (s), 1581 (s), 1502 (s), 1481 (s), 1383 (s), 1267 (s), 1227 (s), 1001 (s), 805 (s). Anal. Calcd for C_41_H_57_N_5_Th: C, 57.80; H, 6.74; N, 8.22. Found: C, 57.78; H, 6.76; N, 8.21. NMR spectroscopy only showed the presence of the resonances of (η^5^-C_5_Me_5_)2_Th[NH(p-tolyl)][κ^2^-C,N-4-(Me_2_N)C_5_H_3_N] (5′) and dmap in C_6_D_6 solution, indicating that an equilibrium between 5 and 5′ + dmap may exist in the solution.
Preparation of (η5-C5Me5)2Th[NH(p-tolyl)](κ2-C,N-1-MeC3H2N2) (6)
Method A
A toluene (5 mL) solution of 1-methylimidazole (21 mg, 0.25 mmol) was added to a toluene (10 mL) solution of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) with stirring at room temperature. After this solution was stirred at room temperature overnight, the solvent was removed under reduced pressure. The residue was extracted with benzene (3 × 10 mL) and then filtered. The volume of the combined filtrate was reduced to 5 mL, and colorless crystals of complex 6 formed upon storage at 10 °C for 2 days. These crystals were isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried under vacuum at room temperature overnight. Yield: 159 mg (92%). M.p.: 60–62 °C (dec.). ^1^H NMR (C_6_D_6): δ 7.13 (d, J = 8.0 Hz, 2H, aryl), 6.95 (m, 3H, aryl), 6.89 (s, 1H, aryl), 4.55 (s, 1H, NH), 3.21 (s, 3H, NCH 3), 2.37 (s, 3H, tolylCH 3), 1.97 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 216.8 (ThC), 155.2 (aryl C), 129.8 (aryl C), 129.6 (aryl C), 123.6 (aryl C), 123.5 (aryl C), 123.2 (aryl C), 118.4 (ring C), 35.7 (NCH_3_), 20.9 (tolylCH_3_), 11.4 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2964 (s), 2911 (s), 1606 (s), 1502 (s), 1397 (s), 1264 (s), 1100 (s), 1021 (s), 805 (s). Anal. Calcd for C_31_H_43_N_3_Th: C, 53.98; H, 6.28; N, 6.09. Found: C, 53.96; H, 6.26; N, 6.12.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of 1-methylimidazole (1.7 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 6 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2Th[NH(p-tolyl)](κ2-C,O-2-CH2–6-MeC5H3NO) (7)
Method A
This compound was prepared as colorless crystals by reacting (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) with 2,6-Me_2_C_5_H_3_NO (31 mg, 0.25 mmol) in toluene (15 mL) at room temperature. Recrystallization from a benzene solution following a procedure analogous to that used in the synthesis of complex 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 157 mg (86%). M.p.: 116–118 °C (dec.). ^1^H NMR (C_6_D_6): δ 7.69 (m, 1H, py), 6.99 (d, J = 8.0 Hz, 2H, phenyl), 6.59 (d, J = 2.2 Hz, 1H, py), 6.49 (d, J = 8.0 Hz, 2H, phenyl), 5.37 (dd, J = 6.6 and 2.6 Hz, 1H, py), 4.96 (s, 1H, NH), 2.28 (s, 2H, CH 2), 2.15 (s, 30H, CpCH 3), 2.08 (s, 3H, CH 3), 1.96 (s, 3H, CH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 161.7 (aryl C), 154.5 (aryl C), 148.2 (aryl C), 129.6 (aryl C), 124.2 (aryl C), 124.0 (aryl C), 119.5 (aryl C), 118.1 (ring C), 113.4 (aryl C), 102.4 (aryl C), 38.0 (ThCH_2_), 27.2 (pyCH_3_), 20.8 (tolylCH_3_), 12.1 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2902 (s), 2856 (s), 1594 (s), 1503 (s), 1433 (s), 1374 (s), 1264 (s), 1137 (s), 1075 (s), 1001 (s), 803 (s). Anal. Calcd for C_34_H_46_N_2_OTh: C, 55.88; H, 6.34; N, 3.83. Found: C, 55.86; H, 7.36; N, 3.81.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of 2,6-Me_2_C_5_H_3_NO (2.5 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 7 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2Th[NH(p-tolyl)][κ2-C,N-N(p-tolyl)P(Me2)CH2] (8)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and Me_3_PO (23 mg, 0.25 mmol) in toluene (15 mL) at room temperature. Recrystallization from a benzene solution following a procedure analogous to that used in the synthesis of complex 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 75 mg (38%; based on Th). M.p.: 116–118 °C (dec.). ^1^H NMR (C_6_D_6): δ 6.97 (d, J = 7.6 Hz, 2H, phenyl), 6.93 (d, J = 8.0 Hz, 2H, phenyl), 6.81 (d, J = 7.6 Hz, 2H, phenyl), 6.16 (d, J = 8.0 Hz, 2H, phenyl), 4.27 (s, 1H, NH), 2.19 (s, 3H, tolylCH 3), 2.15 (s, 3H, tolylCH 3), 2.14 (s, 30H, CpCH 3), 1.03 (d, J P–H = 11.2 Hz, 6H, P(CH 3)2), 0.33 (d, J P–H = 9.5 Hz, 2H, CH 2) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 146.0 (PCH_2_), 132.9 (phenyl C), 129.9 (phenyl C), 129.6 (phenyl C), 128.8 (phenyl C), 128.5 (phenyl C), 127.7 (phenyl C), 125.1 (ring C), 122.9 (phenyl C), 119.1 (phenyl C), 20.7 (d, J P–C = 46.2 Hz, P(CH_3_)2), 19.1 (tolylCH_3_), 18.7 (tolylCH_3_), 12.5 (CpCH_3_) ppm. ^31^P{^1^H} NMR (C_6_D_6_): δ 25.4 ppm. IR (KBr, cm^–1^): ν 2924 (s), 1603 (s), 1380 (s), 1223 (s), 1105 (s), 991 (s), 947 (s), 806 (s). Anal. Calcd for C_37_H_53_N_2_PTh: C, 56.34; H, 6.77; N, 3.55. Found: C, 56.36; H, 6.76; N, 3.57. After isolation of the colorless crystals of 8, the solvent of the mother liquid was removed. ^1^H NMR spectroscopy showed the presence of the resonances of 8, dmap, and other unidentified compounds in the residue.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of Me_3_PN(p-tolyl) (3.6 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 8 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2Th[N(p-tolyl)SS](dmap) (9)
Method A
Solid S_8_ (16 mg, 0.0625 mmol) was added to a toluene (15 mL) solution of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) with stirring at room temperature. After this solution was stirred at 40 °C for 2 days, the solvent was removed. The residue was extracted with benzene (3 × 10 mL) and filtered. The volume of the filtrate was reduced to 5 mL, and colorless microcrystals of 9 formed when this solution was kept at 10 °C for 2 days. Microcrystals of 9 were isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 163 mg (82%). M.p.: 120–122 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.40 (s, 2H, py), 7.68 (d, J = 8.2 Hz, 2H, phenyl), 7.03 (d, J = 8.2 Hz, 2H, phenyl), 6.03 (s, 2H, py), 2.16 (s, 6H, N(CH 3)2), 2.09 (s, 30H, CpCH 3), 1.99 (s, 3H, tolylCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 154.5 (py C), 150.3 (py C), 129.9 (phenyl C), 125.9 (phenyl C), 124.0 (phenyl C), 123.8 (phenyl C), 118.2 (ring C), 106.9 (py C), 38.1 (NCH_3_), 20.8 (tolylCH_3_), 11.9 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2903 (s), 2854 (s), 1597 (s), 1436 (s), 1377 (s), 1088 (s), 1018 (s), 801(s). Anal. Calcd for C_34_H_47_N_3_S_2_Th: C, 51.44; H, 5.97; N, 5.29. Found: C, 51.46; H, 5.96; N, 6.32. Colorless crystals of 9·0.5C_6_H_6_·0.5C_7_H_8_ suitable for X-ray structural analysis were isolated from a mixture of benzene and toluene (4:1) solution.
Method B
NMR Scale
S_8_ (1.3 mg, 0.0051 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.5 mL). Resonances of 9 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 40 °C for 2 days.
Preparation of (η5-C5Me5)2Th[N(p-tolyl)S4] (10)
Method A
This compound was prepared as yellow crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and S_8 (32 mg, 0.125 mmol) in toluene (15 mL) at 40 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 9. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 147 mg (80%). M.p.: 160–162 °C (dec.). ^1^H NMR (C_6_D_6_): δ 7.26 (d, J = 8.2 Hz, 2H, phenyl), 7.04 (d, J = 8.2 Hz, 2H, phenyl), 2.19 (s, 3H, tolylCH 3), 2.12 (s, 15H, CpCH 3), 1.94 (s, 15H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 152.2 (phenyl C), 132.9 (phenyl C), 129.4 (phenyl C), 126.2 (ring C), 125.8 (ring C), 122.7 (phenyl C), 20.9 (tolylCH_3_), 12.1 (CpCH_3_), 12.0 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2962 (s), 2908 (s), 2856 (s), 1614 (s), 1535 (s), 1508 (s), 1442 (s), 1381 (s), 1228 (s), 1004 (s), 804 (s). Anal. Calcd for C_27_H_37_NS_4_Th: C, 44.07; H, 5.07; N, 1.90. Found: C, 44.06; H, 5.09; N, 1.87.
Method B
NMR Scale
S_8_ (2.6 mg, 0.01 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.5 mL). Resonances of 10 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 40 °C for 2 days.
Method C
NMR Scale
S_8_ (1.3 mg, 0.0051 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_Th[N(p-tolyl)SS](dmap) (9; 17.6 mg, 0.02 mmol) and C_6_D_6 (0.5 mL). Resonances of 10 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 40 °C for 2 days.
Preparation of (η5-C5Me5)2Th[N(p-tolyl)Se4] (11)
Method A
This compound was prepared as orange crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and Se (79 mg, 1.00 mmol) in toluene (15 mL) at 40 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 9. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 194 mg (84%). M.p.: 250–252 °C (dec.). ^1^H NMR (C_6_D_6): δ 7.22 (d, J = 8.2 Hz, 2H, phenyl), 7.02 (d, J = 8.2 Hz, 2H, phenyl), 2.18 (s, 3H, tolylCH 3), 2.14 (s, 15H, CpCH 3), 1.94 (s, 15H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 156.6 (phenyl C), 133.0 (phenyl C), 129.1 (phenyl C), 126.4 (ring C), 125.7 (ring C), 123.4 (phenyl C), 20.9 (tolylCH_3_), 12.7 (CpCH_3_), 12.3 (CpCH_3_) ppm. ^77^Se{^1^H} NMR (C_6_D_6_): δ 603.2, 417.3, 385.1, 265.8 ppm. IR (KBr, cm^–1^): ν 2962 (s), 2908 (s), 2854 (s), 1604 (s), 1519 (s), 1504 (s), 1442 (s), 1381 (s), 1226 (s), 1003 (s), 802 (s). Anal. Calcd for C_27_H_37_NSe_4_Th: C, 35.12; H, 4.04; N, 1.52. Found: C, 35.15; H, 4.02; N, 1.54.
Method B
NMR Scale
Se (6.3 mg, 0.08 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.5 mL). Resonances of 11 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 40 °C for 2 days.
Preparation of (η5-C5Me5)2Th[NH(p-tolyl)][κ2-N,Se-2-Se-4-(Me2N)C5H3N] (12)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and Se (20 mg, 0.25 mmol) in toluene (15 mL) at 15 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 9. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 162 mg (80%). M.p.: 218–220 °C (dec.). ^1^H NMR (C_6_D_6): δ 7.70 (d, J = 6.8 Hz, 1H, py), 6.99 (d, J = 8.0 Hz, 2H, phenyl), 6.60 (d, J = 2.5 Hz, 1H, py), 6.49 (d, J = 8.0 Hz, 2H, phenyl), 5.37 (dd, J = 6.7 and 2.5 Hz, 1H, py), 4.96 (s, 1H, NH), 2.28 (s, 3H, tolylCH 3), 2.15 (s, 30H, CpCH 3), 1.96 (s, 6H, N(CH 3)2) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 161.7 (py C), 154.5 (py C), 148.2 (py C), 129.6 (phenyl C), 129.3 (phenyl C), 129.1 (phenyl C), 124.2 (phenyl C), 118.1 (ring C), 113.4 (py C), 102.3 (py C), 38.0 (NCH_3_), 20.8 (tolylCH_3_), 12.1 (CpCH_3_) ppm. ^77^Se{^1^H} NMR (C_6_D_6_): δ 461.1 ppm. IR (KBr, cm^–1^): ν 2902 (s), 1594 (s), 1432 (s), 1384 (s), 1263 (s), 1075 (s), 1001 (s), 800 (s). Anal. Calcd for C_34_H_47_N_3_SeTh: C, 50.49; H, 5.86; N, 5.20. Found: C, 50.47; H, 5.88; N, 5.22.
Method B
NMR Scale
Se (1.6 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.5 mL). Resonances of 12 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 15 °C for 2 weeks.
Preparation of (η5-C5Me5)2Th[NH(p-tolyl)][κ2-N,Te-2-Te-4-(Me2N)C5H3N] (13)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and Te (32 mg, 0.25 mmol) in toluene (15 mL) at reflux and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 9. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 180 mg (84%). M.p.: 180–182 °C (dec.). ^1^H NMR (C_6_D_6): δ 7.85 (d, J = 6.7 Hz, 1H, py), 6.98 (d, J = 8.0 Hz, 2H, phenyl), 6.85 (d, J = 2.7 Hz, 1H, py), 6.46 (d, J = 8.0 Hz, 2H, phenyl), 5.37 (dd, J = 6.7 and 2.7 Hz, 1H, py), 5.01 (s, 1H, NH), 2.26 (s, 3H, tolylCH 3), 2.18 (s, 30H, CpCH 3), 1.88 (s, 6H, N(CH 3)2) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 155.6 (py C), 154.4 (py C), 153.3 (py C), 149.6 (phenyl C), 145.3 (phenyl C), 129.7 (phenyl C), 124.7 (phenyl C), 119.8 (ring C), 118.2 (py C), 103.3 (py C), 37.9 (NCH_3_), 20.8 (tolylCH_3_), 12.8 (CpCH_3_) ppm. ^125^Te{^1^H} NMR (C_6_D_6_): δ 818.1 ppm. IR (KBr, cm^–1^): ν 2901 (s), 2858 (s), 1589 (s), 1502 (s), 1438 (s), 1374 (s), 1262 (s), 1133 (s), 1069 (s), 998 (s), 811 (s). Anal. Calcd for C_34_H_47_N_3_TeTh: C, 47.63; H, 5.53; N, 4.90. Found: C, 47.65; H, 5.51; N, 4.92.
Method B
NMR Scale
Te (2.6 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.5 mL). Resonances of 13 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 120 °C overnight.
Preparation of (η5-C5Me5)2ThF2(dmap) (14)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and AgF (64 mg, 0.50 mmol) in refluxing toluene (15 mL) and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 9. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 146 mg (88%). M.p.: 120–122 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.84 (s, 2H, py), 5.86 (d, J = 5.8 Hz, 2H, py), 2.18 (s, 30H, CpCH 3), 1.99 (s, 6H, NCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 150.8 (py C), 122.1 (ring C), 106.2 (py C), 38.0 (NCH_3_), 10.9 (CpCH_3_) ppm; one carbon of dmap was not observed. IR (KBr, cm^–1^): ν 2962 (s), 2922 (s), 1606 (s), 1523 (s), 1396 (s), 1261 (s), 1091 (s), 1024 (s), 802 (s). Anal. Calcd for C_27_H_40_N_2_F_2_Th: C, 48.94; H, 6.08; N, 4.23. Found: C, 48.95; H, 6.11; N, 4.22.
Method B
NMR Scale
AgF (5.1 mg, 0.04 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.5 mL). Resonances of 14 and those of dmap and other unidentified compounds were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 120 °C overnight.
Preparation of (η5-C5Me5)2ThCl2(dmap)2 (15)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and CuCl (50 mg, 0.50 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 9. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 172 mg (84%). M.p.: 166–168 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.66 (d, J = 6.5 Hz, 4H, py), 5.92 (d, J = 6.2 Hz, 4H, py), 2.30 (s, 30H, CpCH 3), 2.11 (s, 12H, N(CH 3)2) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 154.1 (py C), 151.0 (py C), 126.2 (ring C), 106.2 (py C), 38.1 (NCH_3_), 12.6 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2964 (s), 2933 (s), 2887 (s), 1605 (s), 1527 (s), 1380 (s), 1222 (s), 803 (s). Anal. Calcd for C_34_H_50_N_4_Cl_2_Th: C, 49.94; H, 6.16; N, 6.85. Found: C, 49.95; H, 6.13; N, 6.82.
Method B
NMR Scale
CuCl (4.0 mg, 0.04 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.5 mL). Resonances of 15 and those of dmap and other unidentified compounds were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2ThBr2(dmap)·2C6H6 (16·2C6H6)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and CuBr (72 mg, 0.50 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 9. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 202 mg (86%). M.p.: 146–148 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.55 (s, 2H, py), 7.15 (s, 12H, C_6_ H 6), 5.79 (s, 2H, py), 2.31 (s, 30H, CpCH 3), 2.03 (s, 6H, N(CH 3)2) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 154.2 (py C), 150.4 (py C), 128.5 (C 6_H_6),127.6 (ring C), 105.8 (py C), 38.1 (NCH_3_), 13.2 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2908 (s), 2859 (s), 1616 (s), 1535 (s), 1442 (s), 1384 (s), 1230 (s), 1004 (s), 809 (s). Anal. Calcd for C_39_H_52_N_2_Br_2_Th: C, 49.80; H, 5.57; N, 2.98. Found: C, 49.83; H, 5.55; N, 3.01.
Method B
NMR Scale
CuBr (5.8 mg, 0.04 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.5 mL). Resonances of 16 and those of dmap and other unidentified compounds were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2Th(Cl)[κ2-C,N-4-(Me2N)C5H3N] (17)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and PhSiH_2_Cl (36 mg, 0.25 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 132 mg (80%). M.p.: 208–210 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.11 (d, J = 6.5 Hz, 1H, py), 7.03 (d, J = 2.3 Hz, 1H, py), 5.99 (dd, J = 6.2 and 2.6 Hz, 1H, py), 2.30 (s, 6H, N(CH 3)2), 2.06 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 233.0 (ThC), 154.9 (py C), 143.7 (py C), 123.6 (ring C), 110.4 (py C), 109.1 (py C), 38.7 (NCH_3_), 11.4 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2964 (s), 2932 (s), 1617 (s), 1578 (s), 1431 (s), 1377 (s), 1259 (s), 1019 (s), 989 (s), 803 (s). Anal. Calcd for C_27_H_39_N_2_ClTh: C, 49.20; H, 5.96; N, 4.25. Found: C, 49.22; H, 5.94; N, 4.27.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of PhSiH_2_Cl (2.9 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 17 and those of dmap and p-tolylNHSiH_2_Ph? were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2Th[κ3-C,N,N-(4-Me2NC5H3N)SiH(Ph)N(4-MeC6H3)] (18)
Method A
This compound was prepared as yellow crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and PhSiH_3 (27 mg, 0.25 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 175 mg (84%). M.p.: 286–288 °C (dec.). ^1^H NMR (C_6_D_6_): δ 8.06 (m, 4H, phenyl), 7.31 (t, J = 7.3 Hz, 2H, phenyl), 7.23 (m, 1H, py), 6.98 (m, 1H, phenyl), 6.70 (d, J = 2.9 Hz, 1H, py), 6.64 (d, J = 7.5 Hz, 1H, phenyl), 6.42 (s, 1H, SiH), 6.06 (dd, J = 6.4 and 3.0 Hz, 1H, py), 2.58 (s, 3H, tolylCH 3), 2.04 (s, 15H, CpCH 3), 1.99 (s, 6H, N(CH 3)2), 1.97 (s, 15H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 192.0 (ThC), 168.6 (aryl C), 153.4 (aryl C), 152.9 (aryl C), 147.2 (aryl C), 139.0 (aryl C), 136.2 (aryl C), 135.2 (aryl C), 130.0 (aryl C), 129.3 (aryl C), 128.4 (aryl C), 127.0 (aryl C), 122.3 (ring C), 115.9 (aryl C), 115.0 (aryl C), 104.7 (aryl C), 37.9 (NCH_3_), 22.2 (tolylCH_3_), 11.9 (CpCH_3_), 11.6 (CpCH_3_) ppm. ^29^Si{^1^H} NMR (C_6_D_6_): δ −27.9 ppm. IR (KBr, cm^–1^): ν 2903 (s), 2856 (s), 2066 (s, Si–H), 1598 (s), 1536 (s), 1427 (s), 1378 (s), 1262 (s), 1217 (s), 1001 (s), 968 (s), 818 (s). Anal. Calcd for C_40_H_51_N_3_SiTh: C, 57.61; H, 6.61; N, 5.04. Found: C, 57.62; H, 6.64; N, 5.01.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of PhSiH_3_ (2.2 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 18 and those of dmap and H_2_ were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2Th[κ3-C,N,N-(4-Me2NC5H3N)SiPh2N(4-MeC6H3)]·C6H6 (19·C6H6)
Method A
This compound was prepared as yellow crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and Ph_2_SiH_2 (46 mg, 0.25 mmol) in toluene (15 mL) at 50 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 203 mg (82%). M.p.: 210–212 °C (dec.). ^1^H NMR (C_6_D_6_): δ 8.11 (d, J = 6.3 Hz, 1H, py), 8.07 (s, 1H, phenyl), 8.03 (d, J = 6.5 Hz, 4H, phenyl), 7.23 (m, 6H, phenyl), 7.15 (s, 6H, C_6_ H 6), 6.99 (d, J = 8.0 Hz, 1H, phenyl), 6.90 (m, 2H, phenyl and py), 6.11 (d, 1H, J = 3.6 Hz, py), 2.58 (s, 3H, tolylCH 3), 2.04 (s, 6H, N(CH 3)2), 1.97 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 192.3 (ThC), 169.1 (aryl C), 153.5 (aryl C), 153.3 (aryl C), 147.2 (aryl C), 139.8 (aryl C), 136.0 (aryl C), 129.5 (aryl C), 129.3 (aryl C), 128.53 (aryl C), 128.50 (C 6_H_6), 126.6 (aryl C), 125.6 (aryl C), 122.4 (ring C), 116.7 (aryl C), 115.9 (aryl C), 104.8 (aryl C), 38.0 (NCH_3_), 22.2 (tolylCH_3_), 11.9 (CpCH_3_) ppm. ^29^Si{^1^H} NMR (C_6_D_6_): δ −21.6 ppm. IR (KBr, cm^–1^): ν 2924 (s), 2855 (s), 1597 (s), 1426 (s), 1378 (s), 1263 (s), 1214 (s), 1105 (s), 1002 (s), 808 (s). Anal. Calcd for C_52_H_61_N_3_SiTh: C, 63.20; H, 6.22; N, 4.25. Found: C, 63.22; H, 6.24; N, 4.22.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of Ph_2_SiH_2_ (3.7 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 19 and those of dmap and H_2_ were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 50 °C overnight.
Preparation of (η5-C5Me5)2Th[N(p-tolyl)C(Ph)CHPh][κ2-C,N-4-(Me2N)C5H3N] (20)
Method A
This compound was prepared as yellow crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and PhCCPh (45 mg, 0.25 mmol) in toluene (15 mL) at 80 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 186 mg (82%). M.p.: 180–182 °C (dec.). ^1^H NMR (C_6_D_6): δ 7.83 (d, J = 7.0 Hz, 1H, py), 7.35 (d, J = 7.0 Hz, 2H, phenyl), 7.27 (d, J = 7.3 Hz, 2H, phenyl), 7.00 (m, 3H, phenyl), 6.88 (m, 3H, phenyl), 6.77 (s, 4H, phenyl), 6.46 (d, J = 5.9 Hz, 1H, py), 6.06 (s, 1H, CHC), 5.89 (d, J = 4.0 Hz, 1H, py), 2.31 (s, 6H, N(CH 3)2), 2.22 (s, 3H, tolylCH 3), 2.16 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 232.4 (ThC), 157.6 (aryl C), 154.1 (aryl C), 151.9 (aryl C), 143.6 (aryl C), 141.6 (aryl C), 141.5 (aryl C), 131.9 (aryl C), 130.7 (aryl C), 130.1 (aryl C), 129.3 (aryl C), 128.6 (aryl C), 126.7 (aryl C), 123.6 (ring C), 122.3 (aryl C), 120.4 (aryl C), 117.9 (aryl C), 109.1 (aryl C), 108.0 (CCH), 98.5 (CCH), 38.6 (NCH_3_), 20.8 (tolylCH_3_), 12.2 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2903 (s), 2851 (s), 1578 (s), 1500 (s), 1430 (s), 1364 (s), 1257 (s), 1098 (s), 993 (s), 806 (s). Anal. Calcd for C_48_H_57_N_3_Th: C, 63.49; H, 6.33; N, 4.63. Found: C, 63.52; H, 6.34; N, 4.62.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of PhCCPh (3.6 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 20 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 80 °C overnight.
Preparation of (η5-C5Me5)2Th[N(p-tolyl)C(NC6H11)N(C6H11)](dmap) (21)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and DCC (52 mg, 0.25 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 201 mg (86%). M.p.: 158–160 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.46 (d, J = 6.4 Hz, 2H, py), 7.13 (d, J = 8.2 Hz, 2H, phenyl), 7.03 (d, J = 8.2 Hz, 2H, phenyl), 6.11 (d, J = 6.4 Hz, 2H, py), 4.04 (m, 1H, NCH), 3.70 (m, 1H, NCH), 2.46 (m, 2H, Cy), 2.30 (s, 3H, tolylCH 3), 2.20 (s, 6H, N(CH 3)2), 2.06 (m, 2H, CH_2_), 1.96 (s, 30H, CpCH 3), 1.87 (m, 4H, CH_2_), 1.75 (m, 4H, CH_2_), 1.52 (m, 4H, CH_2_), 1.34 (m, 4H, CH_2_) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 154.1 (py C), 150.5 (py C), 147.3 (phenyl C), 146.7 (phenyl C), 129.4 (phenyl C), 127.3 (phenyl C), 126.4 (ring C), 120.4 (CN), 106.8 (py C), 55.7 (NCH), 54.8 (NCH), 38.2 (NCH_3_), 36.7 (Cy C), 34.9 (Cy C), 27.3 (Cy C), 27.1 (Cy C), 26.6 (Cy C), 25.5 (Cy C), 20.9 (tolylCH_3_), 11.4 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2925 (s), 2852 (s), 1605 (s), 1551 (s), 1506 (s), 1446 (s), 1276 (s), 1230 (s), 1002 (s), 808 (s). Anal. Calcd for C_47_H_69_N_5_Th: C, 60.30; H, 7.43; N, 7.48. Found: C, 60.32; H, 7.44; N, 7.51.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of DCC (4.1 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 21 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2Th[N(p-tolyl)C(N
i Pr)N( i Pr)](dmap) (22)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and DIC (32 mg, 0.25 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 176 mg (82%). M.p.: 114–116 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.46 (d, J = 5.5 Hz, 2H, py), 7.13 (d, J = 7.7 Hz, 2H, phenyl), 7.05 (d, J = 7.7 Hz, 2H, phenyl), 6.10 (d, J = 5.3 Hz, 2H, py), 4.43 (s, 1H, NCH), 4.09 (m, 1H, NCH), 2.31 (s, 3H, tolylCH 3), 2.19 (s, 6H, N(CH 3)2), 1.94 (s, 30H, CpCH 3), 1.60 (d, J = 6.1 Hz, 6H, CH(CH 3)2), 1.41 (d, J = 5.9 Hz, 6H, CH(CH 3)2) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 154.1 (py C), 150.5 (py C), 137.5 (phenyl C), 134.4 (phenyl C), 126.1 (ring C), 123.6 (phenyl C), 122.4 (phenyl C), 118.8 (CN), 106.8 (py C), 51.8 (NCH), 46.7 (NCH), 38.3 (NCH_3_), 26.5 (CH(CH_3_)2), 20.9 (tolylCH_3_), 11.7 (CH(CH_3_)2), 11.2 (CpCH_3_) ppm. IR (KBr, cm^–1^): v 2963 (s), 2919 (s), 2857 (s), 1600 (s), 1541 (s), 1505 (s), 1383 (s), 1262 (s), 1228 (s), 987 (s), 804 (s). Anal. Calcd for C_41_H_61_N_5_Th: C, 57.53; H, 7.18; N, 8.18. Found: C, 57.52; H, 7.15; N, 8.21.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of DIC (2.5 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 22 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2Th[N(p-tolyl)CPh2O](dmap)
(23)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and Ph_2_CO (46 mg, 0.25 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 187 mg (82%). M.p.: 62–64 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.84 (s, 1H, py), 8.44 (s, 1H, py), 8.01 (d, J = 7.5 Hz, 2H, phenyl), 7.90 (d, J = 7.5 Hz, 2H, phenyl), 7.21 (m, 6H, phenyl), 7.10 (m, 4H, phenyl), 6.08 (s, 1H, py), 5.95 (s, 1H, py), 2.35 (s, 3H, tolylCH 3), 2.03 (s, 30H, CpCH 3), 2.01 (s, 6H, N(CH 3)2) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 156.2 (phenyl C), 154.1 (py C), 150.6 (py C), 138.3 (phenyl C), 132.0 (phenyl C), 130.1 (phenyl C), 129.9 (phenyl C), 126.9 (phenyl C), 124.8 (phenyl C), 123.4 (ring C), 121.4 (phenyl C), 106.8 (py C), 92.1 (CO), 38.3 (NCH_3_), 20.7 (tolylCH_3_), 11.8 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2904 (s), 1614 (s), 1537 (s), 1445 (s), 1378 (s), 1226 (s), 1021 (s), 1002 (s), 807 (s). Anal. Calcd for C_47_H_57_N_3_OTh: C, 61.90; H, 6.30; N, 4.61. Found: C, 61.92; H, 6.32; N, 4.59.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of Ph_2_CO (3.6 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 23 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2Th[OCPh2O](dmap)·C6H6 (24·C6H6)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and Ph_2_CO (91 mg, 0.50 mmol) in toluene (15 mL) at 80 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 189 mg (84%). ^1^H NMR (C_6_D_6): δ 9.00 (br s, 2H, py), 8.19 (d, J = 7.7 Hz, 4H, phenyl), 7.30 (t, J = 7.4 Hz, 4H, phenyl), 7.15 (s, 6H, C_6_ H 6), 7.06 (t, J = 6.9 Hz, 2H, phenyl), 6.07 (d, J = 6.0 Hz, 2H, py), 2.08 (s, 6H, N(CH 3)2), 1.93 (s, 30H, CpCH 3) ppm. These spectroscopic data agreed with those reported in the literature.? Furthermore, this complex was also characterized by X-ray diffraction analysis, and its molecular structure is shown in the Supporting Information (Figure S7).
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of Ph_2_CO (7.3 mg, 0.04 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 24 and those of dmap and (p-tolyl)NCPh_2_ ? were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 80 °C overnight.
Preparation of (η5-C5Me5)2Th[SCPh2S](dmap) (25)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and Ph_2_CS (99 mg, 0.50 mmol) in toluene (15 mL) at 50 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 184 mg (86%). M.p.: 128–130 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.74 (d, J = 7.8 Hz, 4H, phenyl), 8.60 (br s, 2H, py), 7.27 (t, J = 7.6 Hz, 4H, phenyl), 7.05 (m, 2H, phenyl), 5.99 (br s, 2H, py), 2.11 (s, 6H, N(CH 3)2), 2.00 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 156.8 (py C), 131.7 (py C), 129.8 (phenyl C), 129.0 (phenyl C), 126.9 (phenyl C), 125.1 (phenyl C), 125.0 (ring C), 106.6 (py C), 58.4 (CS_2_), 38.1 (NCH_3_), 12.5 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2962 (s), 2923 (s), 1607 (s), 1441 (s), 1384 (s), 1260 (s), 1089 (s), 1019 (s), 800 (s). Anal. Calcd for C_40_H_50_N_2_S_2_Th: C, 56.19; H, 5.89; N, 3.28. Found: C, 56.22; H, 5.91; N, 3.26.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of Ph_2_CS (7.9 mg, 0.04 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 25 and those of dmap and (p-tolyl)NCPh_2_ were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 50 °C overnight.
Preparation of (η5-C5Me5)2Th[SCN(p-tolyl)NPh](dmap) (26)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and PhNCS (34 mg, 0.25 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 177 mg (82%). M.p.: 240–242 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.10 (br s, 2H, py), 7.54 (d, J = 7.8 Hz, 2H, tolyl), 7.43 (m, 3H, phenyl), 7.22 (d, J = 7.8 Hz, 2H, tolyl), 7.02 (m, 2H, phenyl), 5.87 (s, 2H, py), 2.11 (s, 3H, tolylCH 3), 2.08 (s, 6H, N(CH 3)2), 1.98 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 154.4 (py C), 149.9 (py C), 137.8 (phenyl C), 129.3 (phenyl C), 125.8 (ring C), 124.6 (phenyl C), 124.5 (phenyl C), 124.4 (phenyl C), 124.3 (phenyl C), 120.7 (phenyl C), 120.6 (phenyl C), 115.4 (CN), 106.7 (py C), 38.1 (NCH_3_), 21.4 (tolylCH_3_), 12.3 (CpCH_3_) ppm. Anal. Calcd for C_41_H_52_N_4_STh: C, 56.93; H, 6.06; N, 6.48. Found: C, 56.92; H, 6.08; N, 6.46.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of PhNCS (2.7 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 26 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of [(η5-C5Me5)2Th]2{μ-[N(p-tolyl)C(S)S]}2·C6H6 (27·C6H6)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and CS_2 (19 mg, 0.25 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 160 mg (84%). M.p.:
300 °C (dec.). ^1^H NMR (C_6_D_6_): δ 7.15 (s, 6H, C_6_ H 6), 7.08 (d, J = 6.9 Hz, 4H, phenyl), 6.98 (d, J = 6.9 Hz, 4H, phenyl), 2.12 (s, 6H, tolylCH 3), 2.06 (s, 30H, CpCH 3), 1.26 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 204.1 (CS_2_), 137.9 (phenyl C), 129.3 (phenyl C), 128.6 (phenyl C), 128.5 (C 6_H_6), 128.2 (phenyl C), 125.6 (ring C), 20.9 (tolylCH_3_), 11.7 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2962 (s), 1590 (m), 1436 (m), 1385 (s), 1260 (s), 1091 (s), 1019 (s), 983 (s), 799 (s). Anal. Calcd for C_68_H_86_N_2_S_4_Th_2_: C, 53.60; H, 5.69; N, 1.84. Found: C, 53.62; H, 5.71; N, 1.86.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of CS_2_ (1.6 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 27 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2Th[N(p-tolyl)CH2C(Me)C(OMe)O](dmap)
(28)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and MMA (36 mg, 0.25 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 170 mg (82%). M.p.: 76–78 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.45 (d, J = 5.4 Hz, 2H, py), 7.13 (d, 2H, J = 8.1 Hz, phenyl), 6.57 (d, 2H, J = 8.1 Hz, phenyl), 6.10 (d, J = 5.8 Hz, 2H, py), 3.96 (s, 2H, CH 2), 3.70 (s, 3H, OCH 3), 2.26 (s, 3H, CH 3), 2.22 (s, 6H, N(CH 3)2), 2.08 (s, 3H, tolylCH 3), 1.93 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 154.1 (py C), 153.6 (phenyl C), 150.6 (py C), 149.7 (phenyl C), 133.3 (phenyl C), 128.6 (phenyl C), 125.6 (phenyl C), 125.1 (ring C), 108.6 (CO), 106.9 (py C), 81.9 (CCO), 53.4 (OCH_3_), 53.1 (CH_2_), 38.3 (NCH_3_), 20.6 (tolylCH_3_), 15.7 (CH_3_), 11.0 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2902 (s), 2856 (s), 1655 (s), 1623 (s), 1545 (s), 1435 (s), 1387 (s), 1261 (s), 1246 (s), 1143 (s), 1000 (s), 807 (s). Anal. Calcd for C_39_H_55_N_3_O_2_Th: C, 56.44; H, 6.68; N, 5.06. Found: C, 56.42; H, 6.71; N, 5.04.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of MMA (2.9 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 28 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of [(η5-C5Me5)2Th]2{μ-[OCH(Me)C(O)OCH(Me)C(N-p-tolyl)O]}2 (29)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and rac-lactide (36 mg, 0.25 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 150 mg (80%). M.p.: 110–112 °C (dec.). ^1^H NMR (C_6_D_6): δ 7.56 (d, J = 8.0 Hz, 4H, phenyl), 7.21 (d, J = 8.0 Hz, 4H, phenyl), 5.71 (m, 2H, OCH), 4.90 (m, 2H, OCH), 2.27 (s, 12H, CH 3), 1.96 (s, 30H, CpCH 3), 1.93 (s, 6H, tolylCH 3), 1.81 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 175.0 (CO_2_), 163.8 (CON), 146.8 (phenyl C), 132.1 (phenyl C), 129.2 (phenyl C), 124.8 (ring C), 124.5 (ring C), 123.1 (phenyl C), 80.3 (OCH), 73.5 (OCH), 30.2 (CH_3_), 23.9 (CH_3_), 20.5 (tolylCH_3_), 11.3 (CpCH_3_), 10.8 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2917 (s), 1676 (s), 1632 (s), 1526 (s), 1404 (s), 1384 (s), 1251 (s), 1151 (s), 1031 (s), 934 (s), 809 (s). Anal. Calcd for C_66_H_90_N_2_O_8_Th_2_: C, 52.72; H, 6.03; N, 1.86. Found: C, 52.74; H, 6.01; N, 1.84.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of rac-lactide (2.9 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 29 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2Th[OC(Ph)N(p-tolyl)]2 (30)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and PhCONH(p-tolyl) (106 mg, 0.50 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 194 mg (84%). M.p.: 170–172 °C (dec.). ^1^H NMR (C_6_D_6): δ 7.64 (d, J = 6.4 Hz, 4H, phenyl), 6.95 (m, 6H, phenyl), 6.47 (s, 8H, phenyl), 2.23 (s, 30H, CpCH 3), 2.08 (s, 6H, tolylCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 174.3 (CO), 145.2 (phenyl C), 136.7 (phenyl C), 132.1 (phenyl C), 130.4 (phenyl C), 130.0 (phenyl C), 129.1 (phenyl C), 128.5 (phenyl C), 125.0 (phenyl C), 123.7 (ring C), 20.9 (tolylCH_3_), 12.1 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2921 (s), 1608 (m), 1508 (s), 1465 (s), 1367 (s), 1265 (s), 1098 (s), 1064 (s), 823 (s). Anal. Calcd for C_48_H_54_N_2_O_2_Th: C, 62.46; H, 5.90; N, 3.04. Found: C, 62.44; H, 5.91; N, 3.06.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of PhCONH(p-tolyl) (8.2 mg, 0.04 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 30 and those of dmap and p-toluidine were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2Th[η3-NHC(Ph)N(p-tolyl)][κ2-C,N-4-(Me2N)C5H3N] (31b)
Method A
This compound was prepared as colorless microcrystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and PhCN (26 mg, 0.25 mmol) in toluene (15 mL) at 80 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 171 mg (82%). M.p.: 158–160 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.03 (d, J = 4.8 Hz, 1H, py), 7.47 (d, J = 7.0 Hz, 2H, phenyl), 6.97 (m, 7H, phenyl), 6.59 (s, 1H, py), 6.09 (s, 1H, py), 6.03 (s, 1H, NH), 2.38 (s, 6H, N(CH 3)2), 2.16 (s, 3H, tolylCH 3), 2.03 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 172.7 (ThC), 154.2 (aryl C), 150.6 (aryl C), 149.3 (aryl C), 142.7 (aryl C), 139.5 (aryl C), 130.3 (aryl C), 129.9 (aryl C), 128.9 (aryl C), 126.5 (aryl C), 123.9 (aryl C), 120.8 (ring C), 111.5 (aryl C), 106.8 (aryl C), 106.6 (CN), 38.6 (NCH_3_), 38.3 (NCH_3_), 20.8 (tolylCH_3_), 11.8 (CpCH_3_), 11.7 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 3395 (m), 2963 (s), 1604 (s), 1523 (s), 1447 (s), 1369 (s), 1260 (s), 1098 (s), 1018 (s), 800 (s). Anal. Calcd for C_41_H_52_N_4_Th: C, 59.12; H, 6.29; N, 6.73. Found: C, 59.14; H, 6.31; N, 6.71. While colorless crystals of (η^5^-C_5_Me_5_)2_Th[N(p-tolyl)C(Ph)N](dmap)·3C_6_H_6 (31a·3C_6_H_6_) and 31b suitable for X-ray structural analysis were isolated from a mixture of benzene and n-hexane (4:1) solution, NMR spectroscopy only showed the presence of the resonances of 31b in C_6_D_6_ solution, indicating an equilibrium between 31a and 31b may exist in the solution.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of PhCN (2.1 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 31b and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 80 °C overnight.
Preparation of (η5-C5Me5)2Th[η7-N(p-tolyl)C(Ph)NC(Ph)NC(Ph)N]·C6H6 (32·C6H6)
Method A
This compound was prepared as red crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and PhCN (78 mg, 0.75 mmol) in toluene (15 mL) at 80 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 209 mg (84%). M.p.: 120–122 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.94 (d, J = 7.2 Hz, 2H, phenyl), 7.60 (d, J = 7.3 Hz, 2H, phenyl), 7.51 (t, J = 7.6 Hz, 2H, phenyl), 7.32 (t, J = 7.3 Hz, 1H, phenyl), 7.15 (s, 6H, C_6_ H 6), 6.83 (m, 6H, phenyl), 6.79 (m, 3H, phenyl), 6.63 (m, 3H, phenyl), 2.12 (s, 30H, CpCH 3), 2.03 (s, 3H, tolylCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 174.5 (ThNC), 158.5 (phenyl C), 156.3 (phenyl C), 150.6 (phenyl C), 143.3 (phenyl C), 141.8 (phenyl C), 141.6 (phenyl C), 134.9 (phenyl C), 133.7 (phenyl C), 129.87 (phenyl C), 129.86 (phenyl C), 129.82 (phenyl C), 129.4 (phenyl C), 129.3 (phenyl C), 129.2 (phenyl C), 128.5 (C 6_H_6), 128.1 (phenyl C), 127.9 (phenyl C), 127.3 (CN), 125.7 (CN), 123.9 (ring C), 20.7 (tolylCH_3_), 11.7 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2915 (m), 1589 (m), 1521 (s), 1446 (m), 1367 (s), 1027 (m), 743 (s). Anal. Calcd for C_54_H_58_N_4_Th: C, 65.18; H, 5.87; N, 5.63. Found: C, 65.15; H, 5.89; N, 5.61.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of PhCN (6.2 mg, 0.06 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 32 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 80 °C overnight.
Preparation of (η5-C5Me5)2Th[η3-N(p-tolyl)C(CH2Ph)NH](NCCHPh) (33)
Method A
This compound was prepared as yellow crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and PhCH_2_CN (59 mg, 0.50 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 173 mg (82%). M.p.: 96–98 °C (dec.). ^1^H NMR (C_6_D_6): δ 7.23 (t, J = 7.4 Hz, 1H, phenyl), 7.12 (d, J = 7.2 Hz, 2H, phenyl), 7.04 (m, 7H, phenyl), 6.95 (d, J = 7.8 Hz, 2H, phenyl), 6.89 (d, J = 7.0 Hz, 2H, phenyl), 5.41 (s, 1H, CCH), 3.46 (s, 1H, NH), 3.40 (s, 2H, CH 2), 2.15 (s, 3H, tolylCH 3), 1.92 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 175.1(CNH), 162.9 (CN), 144.1 (phenyl C), 142.9 (phenyl C), 135.0 (phenyl C), 133.5 (phenyl C), 130.3 (phenyl C), 129.9 (phenyl C), 129.1 (phenyl C), 128.6 (phenyl C), 127.6 (phenyl C), 125.2 (phenyl C), 124.1 (ring C), 122.4 (phenyl C), 122.3 (phenyl C), 118.4 (HCC), 41.2 (CH_2_), 20.9 (tolylCH_3_), 11.2 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 3383 (s, NH), 2908 (s), 2860 (s), 2049 (s, CCN), 1591 (s), 1506 (s), 1296 (s), 1268 (s), 989 (s), 807(s). Anal. Calcd for C_43_H_51_N_3_Th: C, 61.34; H, 6.11; N, 4.99. Found: C, 61.35; H, 6.09; N, 4.97.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of PhCH_2_CN (4.7 mg, 0.04 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 33 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2Th[η3-N(p-tolyl)C(CHPh2)NH](NCCPh2) (34)
Method A
This compound was prepared as yellow crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and Ph_2_CHCN (97 mg, 0.50 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 214 mg (86%). M.p.: 180–182 °C (dec.). ^1^H NMR (C_6_D_6): δ 7.36 (d, J = 7.9 Hz, 4H, phenyl), 7.16 (d, J = 8.0 Hz, 4H, phenyl), 7.07 (m, 5H, phenyl), 7.03 (t, J = 7.2 Hz, 4H, phenyl), 6.94 (m, 5H, phenyl), 6.80 (d, J = 7.2 Hz, 2H, phenyl), 5.74 (s, 1H, NH), 5.21 (s, 1H, CH), 2.13 (s, 3H, tolylCH 3), 1.94 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 176.3 (CNH), 161.6 (CN), 150.5 (phenyl C), 144.0 (phenyl C), 140.7 (phenyl C), 139.5 (phenyl C), 134.1 (phenyl C), 129.7 (phenyl C), 129.6 (phenyl C), 129.2 (phenyl C), 128.9 (phenyl C), 127.6 (phenyl C), 126.5 (phenyl C), 125.8 (phenyl C), 124.8 (ring C), 121.6 (Ph_2_ CC), 42.5 (CH), 20.9 (tolylCH_3_), 11.5 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2970 (s), 2939 (s), 2027 (s), 1635 (s), 1566 (s), 1381 (s), 1265 (s), 1018 (s), 802 (s). Anal. Calcd for C_55_H_59_N_3_Th: C, 66.45; H, 5.98; N, 4.23. Found: C, 66.43; H, 6.01; N, 4.25.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of Ph_2_CHCN (7.7 mg, 0.04 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 34 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature overnight.
Preparation of (η5-C5Me5)2Th[N(p-tolyl)CHN(2,6-Me2C6H3)][κ2-C,N-4-(Me2N)C5H3N]·0.5C6H6 (35·0.5C6H6)
Method A
This compound was prepared as yellow crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and 2,6-Me_2_C_6_H_3_NC (33 mg, 0.25 mmol) in toluene (15 mL) at 40 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 185 mg (82%). M.p.: 228–230 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.73 (s, 1H, CHN), 7.15 (s, 3H, C_6_ H 6), 7.12–6.98 (m, 7H, phenyl and py), 6.91 (d, J = 8.0 Hz, 1H, phenyl), 6.53 (d, J = 8.0 Hz, 1H, py), 5.94 (d, J = 3.8 Hz, 1H, py), 2.35 (s, 6H, N(CH 3)2), 2.28 (s, 3H, tolylCH 3), 2.17 (s, 6H, arylCH 3), 2.07 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 239.3 (ThC), 166.5 (CHN), 162.9 (aryl C), 154.2 (aryl C), 150.6 (aryl C), 150.3 (aryl C), 143.3 (aryl C), 132.6 (aryl C), 131.1 (aryl C), 129.8 (aryl C), 129.3 (aryl C), 128.5 (C 6_H_6), 125.6 (aryl C), 123.0 (ring C), 117.7 (aryl C), 107.4 (aryl C), 38.6 (NCH_3_), 20.9 (tolylCH_3_), 18.5 (arylCH_3_), 12.4 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2913 (s), 2855 (s), 1672 (s), 1600 (s), 1536 (s), 1506 (s), 1433 (s), 1377 (s), 1305 (s), 1193 (s), 993 (s), 806 (s). Anal. Calcd for C_46_H_59_N_4_Th: C, 61.39; H, 6.61; N, 6.23. Found: C, 61.41; H, 6.59; N, 6.25.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of 2,6-Me_2_C_6_H_3_NC (2.6 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 35 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 40 °C overnight.
Preparation of (η5-C5Me5)2Th[NH(p-tolyl)][κ2-C,N-4-(Me2N)-6-(2,6-Me2C6H3NCH)C5H2N] (36)
Method A
This compound was prepared as yellow crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and 2,6-Me_2_C_6_H_3_NC (33 mg, 0.25 mmol) in toluene (15 mL) at 15 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 181 mg (84%). M.p.: 214–216 °C (dec.). ^1^H NMR (C_6_D_6): δ 7.91 (s, 1H, CHN), 7.24 (d, J = 1.8 Hz, 1H, py), 7.06 (m, 2H, aryl), 6.99 (m, 2H, aryl), 6.91 (d, J = 7.8 Hz, 2H, phenyl), 6.53 (d, J = 7.8 Hz, 2H, phenyl), 6.48 (s, 1H, NH), 2.42 (s, 6H, N(CH 3)2), 2.23 (s, 6H, arylCH 3), 2.22 (s, 3H, tolylCH 3), 2.04 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 239.3 (ThC), 162.9 (CHN), 154.9 (aryl C), 154.6 (aryl C), 151.2 (aryl C), 150.9 (aryl C), 129.3 (aryl C), 129.2 (aryl C), 127.0 (aryl C), 125.6 (aryl C), 124.3 (aryl C), 122.6 (ring C), 117.7 (aryl C), 112.0 (aryl C), 110.3 (aryl C), 38.7 (NCH_3_), 20.8 (tolylCH_3_), 18.5 (arylCH_3_), 11.5 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 3219 (m), 1918 (s), 1647 (s), 1599 (s), 1541 (s), 1507 (s), 1432 (s), 1382 (s), 1262 (s), 1089 (s), 1007 (s), 811 (s). Anal. Calcd for C_43_H_56_N_4_Th: C, 59.99; H, 6.56; N, 6.51. Found: C, 60.01; H, 6.54; N, 6.53.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of 2,6-Me_2_C_6_H_3_NC (2.6 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 36 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 15 °C for 1 week.
Preparation of (η5-C5Me5)2Th[NH(p-tolyl)][κ2-C,N-2-(Me3CNC)-4-(Me2N)C5H3N] (37)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and Me_3_CNC (21 mg, 0.25 mmol) in toluene (15 mL) at 50 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 167 mg (82%). M.p.: 160–162 °C (dec.). ^1^H NMR (C_6_D_6): δ 8.38 (d, J = 5.9 Hz, 1H, py), 7.16 (d, J = 8.2 Hz, 2H, phenyl), 6.85 (d, J = 8.2 Hz, 2H, phenyl), 6.51 (d, J = 2.5 Hz, 1H, py), 5.93 (m, 1H, py), 5.37 (s, 1H, NH), 2.42 (s, 6H, N(CH 3)2), 2.36 (s, 3H, tolylCH 3), 2.16 (s, 30H, CpCH 3), 1.47 (s, 9H, C(CH 3)3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 271.5 (ThC), 166.1 (py C), 153.6 (py C), 150.6 (py C), 149.3 (phenyl C), 129.4 (phenyl C), 123.4 (ring C), 118.3 (phenyl C), 106.8 (phenyl C), 104.2 (py C), 102.8 (py C), 64.4 (C(CH_3_)), 38.5 (NCH_3_), 31.8 (C(CH_3_)3), 20.9 (tolylCH_3_), 11.8 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2962 (s), 2908 (s), 2862 (s), 1604 (s), 1512 (s), 1442 (s), 1373 (s), 1226 (s), 1003 (s), 810 (s). Anal. Calcd for C_39_H_56_N_4_Th: C, 57.62; H, 6.94; N, 6.89. Found: C, 57.61; H, 6.96; N, 6.91.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of Me_3_CNC (1.7 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 37 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 50 °C overnight.
Preparation of (η5-C5Me5)2Th[NH(p-tolyl)][κ2-C,N-2-(C6H11NC)-4-(Me2N)C5H3N] (38)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and C_6_H_11_NC (28 mg, 0.25 mmol) in toluene (15 mL) at 15 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 164 mg (78%). M.p.: 178–180 °C (dec.). ^1^H NMR (C_6_D_6): δ 7.19 (d, J = 8.1 Hz, 2H, phenyl), 7.154 (s, 1H, py), 6.91 (d, J = 8.1 Hz, 2H, phenyl), 6.70 (d, J = 2.6 Hz, 1H, py), 5.98 (m, 1H, py), 5.85 (s, 1H, NH), 4.60 (m, 1H, CH), 2.43 (s, 6H, N(CH 3)2), 2,38 (s, 3H, tolylCH 3), 2.18 (s, 30H, CpCH 3), 1.96 (m, 2H, CH 2), 1.67 (m, 2H, CH 2), 1.50 (m, 2H, CH 2), 1.26 (m, 2H, CH 2), 1.13 (m, 2H, CH 2) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 269.9 (ThC), 162.1 (py C), 154.2 (py C), 154.1 (py C), 150.6 (phenyl C), 129.5 (phenyl C), 128.3 (phenyl C), 123.4 (ring C), 118.2 (phenyl C), 106.2 (py C), 105.0 (py C), 63.7 (Cy C), 38.5 (NCH_3_), 35.1 (Cy C), 26.1 (Cy C), 25.6 (Cy C), 20.9 (tolylCH_3_), 11.9 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2928 (s), 1599 (s), 1539 (s), 1518 (s), 1446 (s), 1381 (s), 1226 (s), 987 (s), 804 (s). Anal. Calcd for C_41_H_58_N_4_Th: C, 58.70; H, 6.97; N, 6.68. Found: C, 58.71; H, 6.94; N, 6.71.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of C_6_H_11_NC (2.2 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 38 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 15 °C for 1 week.
Preparation of (η5-C5Me5)2Th[N(p-tolyl)C(CHNCHPh)N(CH2Ph)](dmap) (39)
A benzene (5 mL) solution of PhCH_2_NC (59 mg, 0.50 mmol) was added to a benzene (10 mL) solution of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) without stirring at room temperature. After the solution was stored at 40 °C overnight without stirring, orange crystals were isolated from the solution by filtration. The product was washed with n-hexane (2 mL) and dried at room temperature under vacuum overnight, and the product was identified as 39 by X-ray diffraction analysis. Yield: 202 mg (84%). M.p.: 120–122 °C (dec.). IR (KBr, cm^–1^): ν 2908 (s), 2862 (s), 1597 (s), 1496 (s), 1427 (s), 1288 (s), 810(s). Anal. Calcd for C_50_H_61_N_5_Th: C, 62.29; H, 6.38; N, 7.26. Found: C, 62.27; H, 6.35; N, 7.28. This compound was insoluble in common (deuterated) solvents such as pyridine, THF, toluene, and CD_2_Cl_2, which prevented its characterization by NMR spectroscopy.
Preparation of (η5-C5Me5)2Th[N(p-tolyl)NNN(p-tolyl)] (40)
Method A
This compound was prepared as orange microcrystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and p-tolylN_3 (34 mg, 0.25 mmol) in toluene (15 mL) at 40 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 152 mg (82%). ^1^H NMR (C_6_D_6_): δ 7.12 (d, J = 7.2 Hz, 4H, phenyl), 7.04 (d, J = 7.2 Hz, 4H, phenyl), 2.25 (s, 6H, tolylCH 3), 1.88 (s, 30H, CpCH 3) ppm. These spectroscopic data agreed with those reported in the literature. ?,?
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of p-tolylN_3_ (2.7 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 40 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 40 °C overnight.
Preparation of (η5-C5Me5)2Th[NH(p-tolyl)][κ2-N,N-2-N(NN-p-tolyl)-4-(Me2N)C5H3N] (41)
Method A
This compound was prepared as yellow crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and p-tolylN_3 (34 mg, 0.25 mmol) in toluene (15 mL) at 15 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 168 mg (78%). M.p.: 160–162 °C (dec.). ^1^H NMR (C_6_D_6_): δ 8.00 (d, J = 8.2 Hz, 2H, phenyl), 7.69 (d, J = 6.6 Hz, 1H, py), 7.26 (d, J = 7.9 Hz, 2H, phenyl), 7.15 (d, J = 7.9 Hz, 2H, phenyl), 6.81 (d, J = 8.2 Hz, 2H, phenyl), 6.74 (d, J = 2.5 Hz, 1H, py), 5.36 (dd, J = 6.6 and 2.5 Hz, 1H, py), 5.16 (s, 1H, NH), 2.38 (s, 3H, tolylCH 3), 2.22 (s, 3H, tolylCH 3), 2.19 (s, 6H, N(CH 3)2), 2.15 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 168.1 (py C), 157.1 (py C), 154.9 (py C), 147.0 (phenyl C), 135.8 (phenyl C), 130.0 (phenyl C), 129.5 (phenyl C), 128.5 (phenyl C), 124.9 (ring C), 123.9 (phenyl C), 121.4 (phenyl C), 118.9 (phenyl C), 100.1 (py C), 87.1 (py C), 38.4 (NCH_3_), 21.1 (tolylCH_3_), 20.8 (tolylCH_3_), 11.9 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 2918 (s), 2856 (s), 1602 (s), 1531 (s), 1452 (s), 1384 (s),1369 (s), 1302 (s), 1278 (s), 1195 (s), 1151 (s), 993 (s), 823 (s). Anal. Calcd for C_41_H_54_N_6_Th: C, 57.06; H, 6.31; N, 9.74. Found: C, 57.04; H, 6.34; N, 9.73.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of p-tolylN_3_ (2.7 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 41 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 15 °C for 1 week.
Preparation of (η5-C5Me5)2Th[NH(p-tolyl)][κ2-N,N-2-N(NNCPh3)-4-(Me2N)C5H3N] (42)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and Ph_3_CN_3 (72 mg, 0.25 mmol) in toluene (15 mL) at 50 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 208 mg (82%). M.p.: 152–154 °C (dec.). ^1^H NMR (C_6_D_6_): δ 7.63 (d, J = 6.5 Hz, 1H, py), 7.57 (d, J = 7.6 Hz, 6H, phenyl), 7.46 (s, 1H, NH), 7.18 (m, 6H, phenyl), 7.06 (m, 3H, phenyl), 6.91 (d, J = 7.8 Hz, 2H, phenyl), 6.49 (d, J = 7.8 Hz, 2H, phenyl), 6.05 (d, J = 2.0 Hz, 1H, py), 5.80 (m, 1H, py), 2.29 (s, 6H, N(CH 3)2), 2.27 (s, 3H, tolylCH 3), 2.05 (s, 30H, CpCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 168.0 (py C), 156.3 (py C), 153.6 (py C), 146.5 (phenyl C), 145.0 (phenyl C), 131.2 (phenyl C), 128.9 (phenyl C), 128.5 (phenyl C), 126.8 (phenyl C), 124.5 (ring C), 123.8 (phenyl C), 120.2 (phenyl C), 100.1 (py C), 88.1 (py C), 82.0 (NCPh_3_), 38.7 (NCH_3_), 20.8 (tolylCH_3_), 11.8 (CpCH_3_) ppm. IR (KBr, cm^–1^): ν 3164 (m), 2906 (s), 2856 (s), 1604 (s), 1503 (s), 1433 (s), 1376 (s), 1266 (s), 1162 (s), 999 (s), 750 (s). Anal. Calcd for C_53_H_62_N_6_Th: C, 62.71; H, 6.16; N, 8.28. Found: C, 62.73; H, 6.14; N, 8.31.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of Ph_3_CN_3_ (5.7 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 42 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 50 °C overnight.
Preparation of (η5-C5Me5)2Th(N3)[N(p-tolyl)SiMe3] (43)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and Me_3_SiN_3 (29 mg, 0.25 mmol) in toluene (15 mL) at 40 °C and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 151 mg (84%). M.p.: 182–184 °C (dec.). ^1^H NMR (C_6_D_6_): δ 7.00 (d, J = 7.8 Hz, 2H, phenyl), 6.80 (d, J = 8.0 Hz, 2H, phenyl), 2.16 (s, 3H, tolylCH 3), 1.93 (s, 30H, CpCH 3), 0.39 (s, 9H, SiCH 3) ppm. ^13^C{^1^H} NMR (C_6_D_6_): δ 148.7 (phenyl C), 133.2 (phenyl C), 129.9 (phenyl C), 126.3 (phenyl C), 118.9 (ring C), 20.5 (tolylCH_3_), 11.2 (CpCH_3_), 3.4 (SiCH_3_) ppm. ^29^Si{^1^H} NMR (C_6_D_6_): δ −1.76 ppm. IR (KBr, cm^–1^): ν 2912 (s), 2092 (s), 1611 (s), 1516 (s), 1441 (s), 1383 (s), 1242 (s), 1080 (s), 919 (s), 833 (s). Anal. Calcd for C_30_H_46_N_4_SiTh: C, 49.85; H, 6.41; N, 7.75. Found: C, 49.83; H, 6.44; N, 7.73.
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of Me_3_SiN_3_ (2.3 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 43 and those of dmap were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at 40 °C overnight.
Preparation of [(η5-C5Me5)2Th]2(μ-NNNCSiMe3)2 (44)
Method A
This compound was prepared as colorless crystals from the reaction of (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 213 mg, 0.25 mmol) and Me_3_SiCHN_2 (29 mg, 0.25 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as that in the synthesis of 6. The product was isolated by filtration, rapidly washed with cooled n-hexane (2 mL), and dried at room temperature under vacuum overnight. Yield: 120 mg (76%). ^1^H NMR (C_6_D_6_): δ 2.13 (s, 60H, CpCH 3), 0.52 (s, 18H, SiCH 3) ppm. These spectroscopic data agreed with those reported in the literature.? Furthermore, this complex was also characterized by X-ray diffraction analysis, and its molecular structure is shown in the Supporting Information (Figure S11).
Method B
NMR Scale
A C_6_D_6_ (0.3 mL) solution of Me_3_SiCHN_2_ (2.3 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η^5^-C_5_Me_5_)2_ThN(p-tolyl)(dmap)2 (5; 17.0 mg, 0.02 mmol) and C_6_D_6 (0.2 mL). Resonances of 44 and those of dmap and toluene were observed by ^1^H NMR spectroscopy (100% conversion) when this solution was kept at room temperature for 2 days.
X-ray Crystallography
Single-crystal X-ray diffraction measurements were carried out on a Bruker Smart APEX II CCD diffractometer or on a Rigaku Saturn CCD diffractometer using Mο Kα radiation (λ = 0.71073 Å) or Cu Kα radiation (λ = 1.54184 Å). An empirical absorption correction was applied using the SADABS program.? All structures were solved by direct methods and refined by full-matrix least-squares on F ^2^ using the SHELXL program package.? All the hydrogen atoms were geometrically fixed using the riding model. The crystal data and experimental data for 2–39 and 41–44 are summarized in the Supporting Information. Selected bond lengths and angles are listed in Table.
Computational Methods
Calculations were performed with the Gaussian 09 program (G09),? employing the B3PW91 functional, plus a polarizable continuum model (PCM) (denoted as B3PW91-PCM), with a standard 6–31G(d) basis set for the elements C, H and N and a quasi-relativistic 5f-in-valence effective-core potential (ECP60MWB) treatment with 60 electrons in the core region for Th and the corresponding optimized segmented ((14s13p10d8f6g)/[10s9p5d4f3g]) basis set for the valence shells of Th,? to fully optimize the structures of reactants, transition state(s), intermediates, and products, and to also account for the experimental reaction conditions using toluene as a solvent (dielectric constant ε = 2.379). All stationary points were subsequently characterized by vibrational analyses, from which their respective zero-point (vibrational) energy (ZPE) was extracted and used in the relative energy determinations. In addition, frequency calculations were also performed to ensure that the determined structures for reactants, intermediates, products, and transition states resided at minima and first-order saddle points, respectively, on their potential energy hypersurfaces.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1a Fox A. R.Bart S. C.Meyer K.Cummins C. C.Toward uranium catalysts Nature 200845534134910.1038/nature 0737218800133 · doi ↗ · pubmed ↗
- 2a Burns C. J.Smith W. H.Huffman J. C.Sattelberger A. P.Uranium(VI) Organoimido Complexes J. Am. Chem. Soc.19901123237323910.1021/ja 00164 a 069 · doi ↗
- 3a Cramer R. E.Panchanatheswaran K.Gilje J. W.Uranium-Carbon Multiple-Bond Chemistry. 3. Insertion of Acetonitrile and the Formation of a Uranium-Nitrogen Multiple Bond J. Am. Chem. Soc.19841061853185410.1021/ja 00318 a 059 · doi ↗
- 4a Haskel A.Straub T.Eisen M. S.Organoactinide-Catalyzed Intermolecular Hydroamination of Terminal Alkynes Organometallics 1996153773377510.1021/om 960182 z · doi ↗
- 5w Walter, M. D. Organouranium Chemistry in Encyclopedia of Inorganic and Bioinorganic Chemistry, Scott, R. A. , Ed.; John Wiley & Sons, 2023.
- 6a Barros N.Maynau D.Maron L.Eisenstein O.Zi G.Andersen R. A.Single but Stronger UO, Double but Weaker UN Me Bonds: The Tale Told by Cp 2UO and Cp 2UNR Organometallics 2007265059506510.1021/om 700628 e · doi ↗
- 7a Ren W.Zi G.Fang D.-C.Walter M. D.Thorium Oxo and Sulfido Metallocenes: Synthesis, Structure, Reactivity, and Computational Studies J. Am. Chem. Soc.2011133131831319610.1021/ja 205280 k 21793520 · doi ↗ · pubmed ↗
- 8Fagan P. J.Manriquez J. M.Maatta E. A.Seyam A. M.Marks T. J.Synthesis and Properties of Bis(pentamethylcyclopentadienyl) Actinide Hydrocarbyls and Hydrides. A New Class of Highly Reactive f-Element Organometallic Compounds J. Am. Chem. Soc.19811036650666710.1021/ja 00412 a 021 · doi ↗