Karakum desert: a unique source of cultivable novel and rare actinomycetes with a remarkable biosynthetic potential
Hayrettin Saygin, Nevzat Sahin, Michael Goodfellow

TL;DR
Scientists discovered new and rare actinomycete bacteria in the Karakum Desert that can produce valuable antibiotics and plant growth-promoting compounds.
Contribution
The study identifies novel and rare actinomycetes from the Karakum Desert with unique biosynthetic potential for antibiotics and plant growth promotion.
Findings
459 actinomycete isolates were obtained, including representatives from 17 genera, some of which are novel or rare.
Genome analysis of 32 isolates revealed biosynthetic gene clusters for novel metabolites, including antibiotics.
Many isolates contain genes promoting plant growth and adapting to harsh desert conditions.
Abstract
A culture-based strategy was used to isolate and screen representative actinomycetes from six sampling sites in the Karakum Desert, Turkmenistan. A total of 459 actinomycete isolates were obtained using 16 selective media, and 270 representative strains were subjected to 16 S rRNA gene sequencing. Comparative 16 S rRNA gene sequence analyses on colour-group representatives led to their assignment to 17 genera with validly published names which included many isolates assigned to novel or putatively novel species including ones belonging to rare genera, such as Actinocorallia, Actinomadura, Jiangella and Nonomuraea. Mining of whole-genome sequences of 32 isolates which formed distinct lineages in phylogenomic trees revealed biosynthetic gene clusters predicted to encode for many novel specialized metabolites, notably antibiotics. The genomes of most of these isolates included genes…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
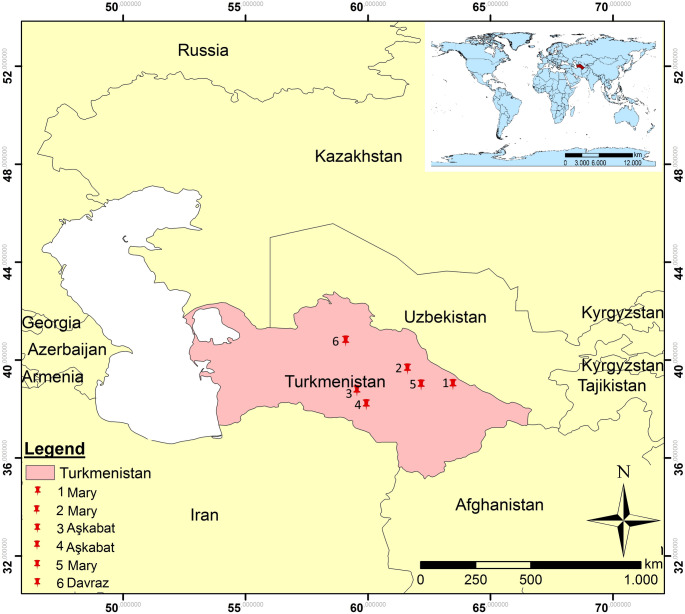 Figure 1
Figure 1 Figure 2
Figure 2 Figure 3
Figure 3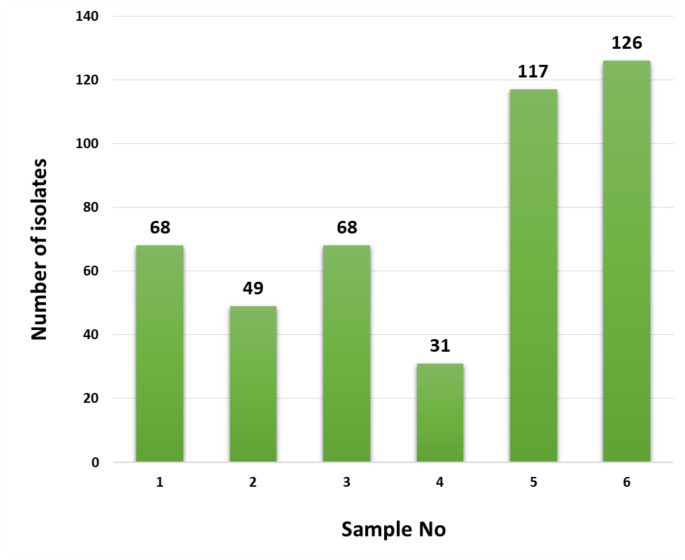 Figure 4
Figure 4 Figure 5
Figure 5 Figure 6
Figure 6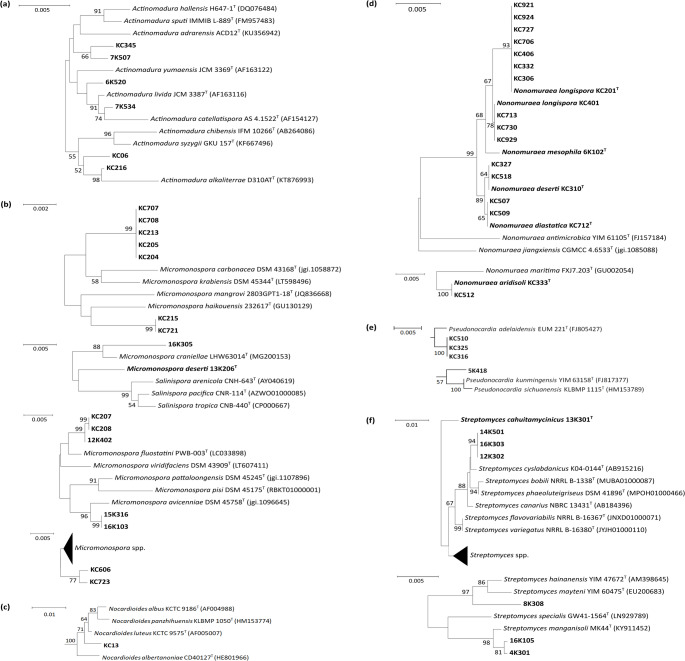 Figure 7
Figure 7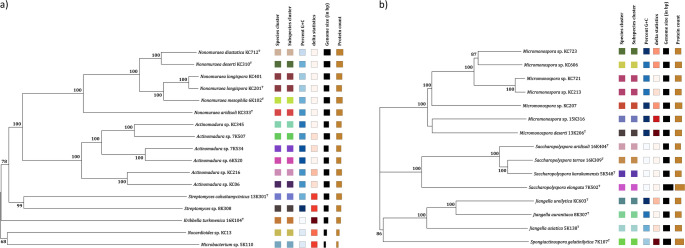 Figure 8
Figure 8- —Ondokuz Mayıs University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMycorrhizal Fungi and Plant Interactions · Microbial Natural Products and Biosynthesis · Plant Pathogens and Fungal Diseases
Introduction
Natural products remain the most prolific source of pharmaceutically interesting biomolecules (Newman and Cragg 2020) needed to control multidrug-resistant microbial pathogens and to treat patients with life-threatening diseases such as cancer (Sivalingam et al. 2019). Filamentous actinomycetes classified in the phylum Actinomycetota (Oren and Garrity 2021) are, and remain, a unique source of novel antibiotics (Takahashi and Nakashima 2018). Streptomycetes are especially gifted sensu Baltz (2017) as they have large genomes rich in natural product – biosynthetic gene clusters (NP – BGCs) many of which encode for new and uncharacterised antibiotics (Sivakala et al. 2021). Other gifted filamentous actinomycetes include ones classified in the genera Amycolatopsis (Sangal et al. 2018). Micromonospora (Carro et al. 2019a), Nocardia (Männle et al. 2020) and Salinispora (Jensen et al. 2015). Rare and novel actinomycetes are an asset in the search for novel bioactive compounds using emerging technologies, notably genome mining, metabolic engineering, metabolomics, proteomics and transcriptomics (Sekurova et al. 2019; Atanasov et al. 2021; Ossai et al. 2022). Questions arising from these developments include “how and where to look for novel gifted actinomycetes”.
Extreme biomes are promising sources of gifted filamentous actinomycetes as harsh abiotic conditions select for novel actinomycetes with unexplored chemical diversity that can lead to the discovery of new bioactive compounds (Rateb et al. 2018; Sayed et al. 2020a). The genomes of some gifted filamentous actinomycetes from extreme habitats contain clade and species-specific NP-BGCs (Adamek et al. 2018; Carro et al. 2018; Nouioui et al. 2019); a development which underpins the importance of sound classification in bioprospecting campaigns, notably taxonomic approaches to drug discovery (Świecimska et al. 2023). Genome-based taxonomies also bring greater precision to bioprospecting and ecological studies (Nouioui et al. 2018; Sangal et al. 2018), as do genomic-based metrics used to delineate generic, species and subspecific boundaries (Meier-Kolthoff et al. 2014; Sant’Anna et al. 2019; Thompson et al. 2020). Further, the detection of stress-related genes in genomes of actinomycetes isolated from extreme habitats provide genomic insights into how they have adapt to extreme environmental conditions (Busarakam et al. 2016; Abdel-Mageed et al. 2020).
Until recently, little was known about the numbers, types and distribution of actinomycetes in deserts even though desert biomes account for about 20% of the Earth’s landmass around 7% of which is hyper-arid (Neilson et al. 2012; Mohammadipanah and Wink 2016). Desert habitats are an especially rich source of novel and rare filamentous actinomycetes that produce new specialized (secondary) metabolites, such as anticancer compounds, antibiotics, immunosuppressive agents and enzyme inhibitors (Xie and Pathom-aree 2021). Small numbers of taxonomically diverse, bioactive, filamentous actinomycetes are characteristic of the Atacama (Goodfellow et al. 2018), Chihuahuan (Arocha-Garza et al. 2017), Kazakhstan (Ziyat et al. 2019), Mongolian (Kurapova et al. 2012), Gurbantunggut (Li et al. 2021), Mojave (McHugh et al. 2017), Taklamakan (Liu et al. 2021), Thar (Masand et al. 2018) and Sahara (Meklat et al. 2020) deserts, polar desert soils (Ji et al. 2022), the desert ecosystem of the Qinghai-Tibet plateau (Ding et al. 2013) and the cold desert of the Leh Ladakh region of Jammu and Kashmir (Yadav et al. 2015).
Actinomycetes from Central and South Asian deserts have rarely featured in biotechnological or ecological studies (Xie and Pathom-aree 2021; Xie et al. 2022). This also applies to the Karakum Desert, one of the largest deserts in the world and the hottest in Central Asia (Ghassemi and Garzanti 2019). Recently, small numbers of filamentous actinomycetes isolated from undisturbed habitats in the Karakum Desert were described as novel Jiangella (Saygin et al. 2020a), Kribbella (Saygin et al. 2019a), Micromonospora (Saygin et al. 2020c), Nonomuraea (Saygin et al. 2020d, 2021a), Saccharopolyspora (Saygin et al. 2021b), Spongiactinospora (Saygin et al. 2019b; Ay et al. 2020) and Streptomyces (Saygin et al. 2020b) species thereby providing additional evidence that neglected desert biomes are a source of gifted filamentous actinomycetes with the potential to produce new drug leads and compounds that promote and protect plant growth (Nouioui et al. 2019; Ebrahimi-Zarandi et al. 2022).
In this study, representative amycelial and filamentous actinomycete colonies from selective isolation plates inoculated with environmental samples from six locations in the Karakum Desert were assigned to colour-groups. 16 S rRNA gene sequence analyses were carried out on representatives of the colour-groups to determine their taxonomic status and the resultant phylogenetic data used to establish the identity of isolates assigned to the colour-groups. Whole-genome sequences generated for 32 isolates that differed markedly from their phylogenetic neighbours were examined for BGCs predicted to express for specialized metabolites, notably antibiotics, for genes which code for compounds that promote plant growth and stress-related genes. The overall objectives of the study were to determine the taxonomic diversity of culturable actinomycetes, the functional activities and genetic potential of representative isolates, and to generate a high quality library of taxonomically diverse actinomycetes that can be used to address critical contemporary issues facing agricultural, industrial and medical biotechnology.
Materials and methods
Karakum desert: geography, biology and sampling sites
The Karakum Desert, which covers about 80% of Turkmenistan (Zonn and Esenov 2014), lies between the River Amudarya in the east and the Caspian Sea in the west (Ghassemi and Garzanti 2019), has extensive underground oil and gas reserves (Arbatov and Karatepe 2006). The region has a seasonal climate, cold winters, warmish dry autumns, gentle rain in spring, and very hot cloudless summers (maximum air temperatures 48 °C to 50 °C in July with ground temperatures above 80 °C (Zonn and Esenov 2014). In July 2014, when the environmental samples were taken, the Karakum Desert had temperatures varying between 23 and 38 °C, a monthly precipitation of about 5 mm, and a wind speed of 1–2 m per second (www.weather-and-climate.com). Environmental samples (Table 1) were collected from selected regions of the Karakum Desert (Figs. 1 and 2). About 200 g of subsurface soil (depth 5 cm) was collected aseptically at each of the pristine sites using a spatula sterilized in the field with ethanol, and placed into sterilized polyethylene bags. The samples were brought to the laboratory quickly and stored at 4 °C.
Table 1. Sampling sitesSample no and locationLatitude °NLongitude °EAltitudeDate of sampling1. Mary38°12’31.41”62°53’37.29”190 m06.07.20142. Mary38°57’18.94”61°29’10.26”154 m06.07.20143. Aşkabat38°25’48.54”58°29’38.60”86 m06.07.20144. Aşkabat38°04’06.25”58°43’10.59”131 m06.07.20145. Mary38°03’25.92”61°50’05.63”198 m06.07.20146. Darvaz40°15’38.13”58°26’20.39”90 m06.07.2014
Fig. 1. Location of the Karakum Desert sampling sites
Fig. 2. Selected sampling locations in the Karakum Desert; (a) Mary (sample no 1), (b) Aşkabat (sample no 3), (c) Mary (sample no 5) and (d) Darvaz (sample no 6)
Physico-chemical analyses of environmental samples
The pH of each of the samples was determined using the procedure of Reed and Cummings (1945) and electrical conductivity measurements after Avery and Bascomb (1982). The moisture content of the samples were determined by drying known amounts to constant weight at 105 °C. The organic matter content of each sample was calculated using the modified Walkley-Black method (Walkley and Black 1934).
Selective isolation, purification and preservation of isolates
Environmental samples from each location were air-dried at room temperature for 14 days, and 1 gram of each soil sample was suspended in 9 mL of sterile ¼ strength Ringer’s solution (Oxoid) and agitated on a rotary shaker at 160 rpm for 30 min to release microbial cells. The resulting suspensions were then held at 60 °C for 20 min, and serial 10^− 2^ to 10^− 3^ dilutions were prepared using the same diluent. Serial dilutions (200 µl) prepared from each of the samples were spread over the surfaces of 16 isolation media considered to be selective for taxonomically diverse actinomycetes (Table 2). The numbers of presumptive actinomycetes growing on the isolation plates were expressed as the number of colony-forming units (cfu) per gram dry weight soil following incubation at 28 °C for 4 weeks.
Table 2. Media used for the selective isolation of actinomycetesMediaSelective agentsTarget organismsReferencesSelective medium 1 (SM1) agarNeomycin (4 µg/ml), nystatin (50 µg/ml), D-sorbitol (10%)Amycolatopsis spp.Tan et al. (2006)Selective medium 2 (SM2) agarNeomycin (4 µg/ml), nystatin (50 µg/ml), D-melezitose (10%)Amycolatopsis spp.Tan et al. (2006)Selective medium 3 (SM3) agarNalidixic acid (10 µg/ml), novobiocin (10 µg/ml)Amycolatopsis spp.Tan et al. (2006)Marine agarNalidixic acid (10 µg/ml)ActinomycetesZoBell (1941)Humic acid-vitamin agarNalidixic acid (10 µg/ml), humic acid (1 g/l)Streptosporangium spp.Hayakawa and Nonomura (1987)Starch-peptone-yeast extract (M1) agarRifampicin (5 µg/ml)Salinispora spp.Mincer et al. (2002)Non-sporulating agarRifampicin (5 µg/ml)Rare actinomycetesSanglier et al. (1992)Reasoner’s 2 A (R2A) agarNalidixic acid (10 µg/ml)Modestobacter spp.Reasoner and Geldreich (1985)Starch-casein agarNalidixic acid (10 µg/ml)Streptomyces spp.Küster and Williams (1964)Raffinose-histidine agarNalidixic acid (10 µg/ml)Rare Streptomyces spp.Vickers et al. (1984)Tryptic soy agarNalidixic acid (10 µg/ml)ActinomycetesDifco (Cat. No 236950)Oligotrophic agarNalidixic acid (10 µg/ml), low carbon and nitrogen contentRare and uncommon actinomycetesJiang et al. (2016)Minimal mediumNalidixic acid (10 µg/ml)Rare and uncommon actinomycetesJiang et al. (2016)Complex mediumNalidixic acid (10 µg/ml)Nocardiopsis spp.Chun et al. (2000)Glycerol-asparagine agarNalidixic acid (10 µg/ml)Streptomyces spp.Shirling and Gottlieb (1966)Chitin-vitamin agarNalidixic acid (10 µg/ml)Streptomyces spp.Hayakawa and Nonomura (1987)All of the media were supplemented with cycloheximide (50 µg/ml)
Presumptive actinomycete colonies taken from each of the selective isolation media were subcultured onto yeast extract-malt extract agar (ISP 2) (Shirling and Gottlieb 1966) plates which were incubated at 28 °C for 14 days. Growth from each of the ISP 2 plates was suspended in 25 % (v/v) glycerol held in vials, and the resultant cultures were preserved at -20 °C and − 80 °C. Colonies were assigned to colour-groups based on aerial spore mass, substrate mycelial, and diffusible pigment colours or colony pigments on oatmeal agar plates (ISP 3) using colour charts (Centore 2016).
Phylogenetic analyses
Isolates representing the colour-groups were shaken individually in ISP 2 broth at 160 rpm for 14 days at 28 °C and the resultant biomass was harvested by centrifugation and washed twice with an equal volume of sterile distilled water. Genomic DNA was extracted using PureLink^®^ DNA Isolation Kits (Invitrogen, USA). PCR-mediated amplification of 16 S rRNA genes was achieved using the universal primers, 27 F and 1525R (Lane 1991). The PCR products were purified and sequenced using an ABI PRISM 3730 XL automatic sequencer with forward primers 27 F (Lane 1991), 518 F (Buchholz-Cleven et al. 1997), MG5F and MG6F (Chun and Goodfellow 1995) and the reverse primer 800R (Chun and Goodfellow 1995).
Chromatogram files in ABI format were checked using Chromas version 1.45, and the primers overlapped to obtain the 16 S rRNA gene nucleotide sequences in FASTA format for each isolate. The resultant 16 S rRNA gene sequences were deposited in GenBank under accession numbers MG770618-…-MG770879 and MK156407-…-MK156414. The 16 S rRNA gene sequences were uploaded onto the EzBioCloud server (Yoon et al. 2017) and pairwise sequence similarities of the nearest phylogenetic neighbours determined. Phylogenetic trees were generated using the MEGA X software package (Kumar et al. 2018) using the neighbour-joining method (Saitou and Nei 1987). The topologies of the resultant trees were evaluated by bootstrap resampling of 1000 replicates after Felsenstein (1985).
Whole-genome sequencing of selected isolates
Thirty-two isolates (Table S1), which differed markedly from the type strains of their closest phylogenetic neighbours, were grown in ISP 2 broth at 160 rpm for 7 days at 28 °C; the biomass was collected by centrifugation and washed twice with an equal volume of sterile distilled water. The genomic DNA of each isolate was extracted using PureLink^®^ DNA Isolation Kits (Invitrogen, USA). The quantity of the DNA preparations was measured using NanoDrop 2000 (Thermo Scientific, USA). The whole-genome sequences of the isolates were generated using the Illumina HiSeq 2500 next-generation sequencing platform and the ×250 bp paired-end protocol. Assemblies of raw data were achieved using the full Spades assemble strategy on the Bacterial and Viral Bioinformatics Resource Center (BV-BRC) (https://www.bv-brc.org/) (Olson et al. 2023), and the draft genome sequences deposited in the National Centre for Biotechnology Information (NCBI) database (https://submit.ncbi.nlm.nih.gov/) database under the accession numbers (Table S1). The genome sequences were annotated using the Rapid Annotation Subsystem Technology (RAST) server (Aziz et al. 2008), and uploaded onto the Type (Strain) Genome Server (TYGS), a free bioinformatics platform available at https://tygs.dsmz.de (Meier-Kolthoff and Göker 2019), to construct a phylogenomic tree. The presence of BGCs in the genomes of the isolates were detected using the antiSMASH v7.0 (Blin et al. 2023), with default options available at https://antismash.secondarymetabolites.org.
Antimicrobial screening of representative isolates
Fifty-two isolates, which differed markedly from their phylogenetic neighbours, were assigned to 11 genera. These strains were examined for their ability to inhibit the growth of a panel of bacteria and fungi, namely Bacillus subtilis ATCC 6633, Escherichia coli ATCC 25,922, Enterococcus faecalis ATCC 29,212, Klebsiella quasipneumoniae ATCC 700,603, Pseudomonas aeruginosa ATCC 27,853, Staphylococcus aureus ATCC 25,923, Aspergillus brasiliensis ATCC 16,404 and Candida albicans ATCC 10,231, using an agar diffusion procedure as described by Balouiri et al. (2016). The resultant preparations were incubated for 24–48 h at optimal temperatures for the wild-type strains, and the activity of the strains was recorded by measuring inhibition zones around the colonies. Inhibition zones with a diameter of ≥ 10 mm, including the well diameter, were considered indicative of antimicrobial activity.
Results
Physico-chemical characteristics of environmental samples
All of the samples were moderately alkaline, highly arid with low levels of organic matter and small electrical conductivity values (Table 3). The samples taken from locations 1 to 4 and 6 had electrical conductivity, moisture, pH and organic matter values that fell within a narrow range, namely 0.12–0.20 (dS/m), 0.20–0.42%, 8.44–8.66 and 0.04–0.20%, respectively. In contrast, the sample from location 5 showed the highest electrical conductivity, and moisture content with the lowest pH and equal lowest total organic matter content.
Table 3. Physico-chemical properties of the environmental samplesSampling siteTotal organic matter (%)pHElectrical conductivity (dS/m)Moisture content (%)10,048,650,140.3220,168,710,120.3630,188,660,130.3740,128,440,140.4250,048,250,750.7260,208,610,200.20
Colony counts, strain selection and assignment of representative isolates to colour-groups
With few exceptions, small numbers of presumptive actinomycete colonies were recorded on the selective isolation plates prepared from environmental samples taken from the sampling locations (Table 4). In all cases the highest counts were recorded from the humic acid-vitamin agar plates with numbers ranging from 55.2 × 10^3^ cfu’s for the sample from location 4 to 217.3 × 10^3^ cfu’s for the sample taken from location 5. In contrast, the lowest counts were found on the three SM media designed to isolate Amycolatopsis strains (Tan et al. 2006) and on the tryptic soy agar (TSA) plates. The highest average actinomycete counts were from the samples collected from locations 5 and 6. Four hundred and fifty nine isolates representing distinctive colony types were taken from each of the isolation plates. Of these, 270 representative strains were selected for 16 S rRNA gene sequencing and phylogenetic analysis. Most of these strains produced filamentous colonies covered by either aerial hyphae or by a characteristic aerial spore mass (Fig. 3); presumptive amycelial actinomycete colonies were selected based on colony pigmentation.
Fig. 3. Isolates growing on (a) humic acid-vitamin agar plates showing colonies covered by white aerial hyphae, (b) M1 agar plates with round colonies carrying a grey aerial spore mass, and (c) raffinose-histidine agar plates showing masses of aerial spores following incubation at 28 °C for 28 days
The most pronounced range of colony types were from the samples collected from locations 5 and 6 (Fig. 4). The representative isolates from the sampling sites were assigned to 88 multiple-membered (2 to 13 isolates) and 35 single-membered colour-groups based on pigmentational properties recorded from the oatmeal agar plates (Table S2). Colony morphologies of a few selected isolates grown on ISP 2 agar, amycelial isolates were distributed to colour-groups based on colony colour (Fig. 5).
Table 4. Total number of actinomycetes (10^3^ Cfu/g dry weight soil) growing on isolation media inoculated with serial dilutions prepared from environmental samples collected from the sampling sites following incubation at 28 °C for four weeksSample Sites123456Total counts Media SM1 agar-1.01.5-3.50.56.5SM2 agar1.5-1.5-3.02.58.5SM3 agar1.51.50.50.51.56.512Marine agar15.512.810.67.922.475.1144.3Humic acid-vitamin agar178.278.5145.555.2217.3128.5803.2M1 agar4.81.64.62.925.429.668.9Non-sporulating agar8.52.34.12.625.54.647.6R2A agar4.85.27.11.55.045.869.4Starch-casein agar28.836.534.642.552.242.0236.6Raffinose-histidine agar7.513.212.82.538.51.075.5Tryptic soy agar2.0--1.53.51.58.5Oligotrophic agar12.54.55.05.46.52.035.9Minimal agar17.517.820.52.03.0-60.8Complex agar1.04.55.51.21.50.514.2Glycerol-asparagine agar4.51.825.510.31.2-43.3Chitin-vitamin agar16.515.826.413.22.13.077 Average counts 19.012.319.19.325.821.4106.9-, Actinomycete colonies were not present on the isolation plates
Fig. 4. Numbers of strains taken to represent the different colony types of actinomycetes from sampling locations 1 to 6
Fig. 5. Representative filamentous actinomycetes growing on yeast extract-malt extract (ISP 2) agar plates showing colonies bearing pigmented spores on aerial mycelia after 14 days growth at 28 °C
Taxonomic diversity of representatives of colour-groups
The 16 S rRNA gene sequence analyses showed that 259 out of the 270 isolates (96%) taken to represent the colour-groups belong to genera classified in the phylum Actinomycetota (Table 5), the remainder were assigned to eleven non-actinomycete taxa (Table S2).
Table 5. Assignment of representative Karakum desert isolates to genera, families and orders (n = 259 isolates) *OrderFamilyGenusIsolate Corynebacteriales
Nocardiaceae
Nocardia 6K504, 6K505, 6K519, 7K517, 7K520, 7K528, KC209, KC320, KC910, KC916 Jiangellales
Jiangellaceae
Jiangella 1K503, 5K116, *****5K138^T^, *****8K307^T^, *****KC603^T^ Microbacteriales
Microbacteriaceae
Agromyces 4K403B Microbacterium *****5K110 Micromonosporales
Micromonosporaceae
Micromonospora 4K205, 5K118, 5K221, 5K409, 5K544, 7K104, 7K504, 7K530, 8K103, 8K503, 10K527, 12K108, 12K402, 13K111, 13K203, *****13K206^T^, 13K208, 15K101, 15K103, 15K303, *****15K316, 16K103, 16K305, 16K405, KC202, KC204, KC205, *****KC207, KC208, *****KC213, KC215, KC605, *****KC606, KC707, KC708, *****KC721, *****KC723 Plantactinospora 5K208, KC601, KC705, KC725, KC728, KC729 Propionibacteriales
Kribbellaceae
Kribbella 7K109, *****16K104^T^ Nocardioidaceae
Nocardioides KC13^T^ Pseudonocardiales
Pseudonocardiaceae
Pseudonocardia 5K133, 5K313, 5K418, KC301, KC304, KC311, KC316, KC323, KC325, KC502, KC504, KC510 Saccharomonospora 6K501 Saccharopolyspora 4K102, 5K506, *****5K548^T^, 6K514, *****7K502^T^, *****16K309^T^, *****16K404^T^, KC101, KC103, KC408 Streptomycetales
Streptomycetaceae
Streptomyces 3K201, 3K203, 3K401, 4K203, 4K301, 4K405, 5K101, 5K103, 5K109, 5K114, 5K122, 5K125, 5K131, 5K132, 5K135, 5K209, 5K312, 5K316, 5K417, 5K505, 5K520, 5K524, 5K526, 5K531, 5K532, 5K551, 6K103, 6K201, 6K502, 6K525, 7K111, 7K201, 7K301, 7K304, 7K305, 7K501, 7K513, 7K522, 7K529, 8K102, 8K205, 8K206, 8K301, 8K308, *****8K508, 10K101, 10K102, 10K208, 10K209, 10K210, 10K212, 10K305, 10K307, 10K402, 10K507, 10K514, 10K516, 11K101, 11K402, 11K502, 12K107, 12K109, 12K203, 12K302, 12K506, 13K103, 13K105, 13K109, 13K204, *****13K301^T^, 13K403, 14K201, 14K203, 14K501, 15K302, 15K408, 16K102, 16K105, 16K107, 16K205, 16K207, 16K210, 16K211, 16K303, 16K306, 16K311, 16K401, 16K501, KC11, KC12, KC15, KC18, KC319, KC340, KC403, KC407, KC410, KC416, KC421, KC426, KC501, KC506, KC903, KC907, KC918, KC930 Streptosporangiales
Nocardiopsaceae
Nocardiopsis 3K402, 5K222, 5K521, 10K510, 15K402, 16K403 Streptosporangiaceae
Nonomuraea 1K501, 4K104, 4K202, 4K403A, 5K134, 5K320, 5K322, *****6K102^T^, 7K110, 7K519, 7K523, 7K532, 8K306, 15K203, 15K301, *****KC201^T^, KC306, *****KC310^T^, KC327, KC332, *****KC333^T^, *****KC401, KC406, KC507, KC509, KC512, KC518, KC706, *****KC712^T^, KC713, KC727, KC730, KC921, KC924, KC929 Spongiactinospora *****13K107^T^ Thermomonosporaceae
Actinocorallia 5K502, 5K516, 5K550 Actinomadura 5K515, 5K555, 6K515, *****6K520, 6K521, 6K523, 7K401, *****7K507, 7K515, 7K525, 7K527, 7K533, *****7K534, 7K537, 10K502, 10K504, 10K511, 10K521, *****KC06, *****KC216, *****KC345, KC608Based on the classification of Salam et al. (2020) *****Isolates included in the phylogenomic analysesThe following codes show the source of the isolates and the selective medium on which they were cultivated (a) The numbers before the “K” code refer to selective isolation medium (i.e. 1–16 as shown in Table 4) and after the “K” code they refer to the environmental sample (i.e. 1–5, as shown in Fig. 1) (first digit) and strain numbers (last two digits), as exemplified by 13K206, isolate 6 from site 2 isolated on minimal medium agar; (b) “KC” refers to isolates recovered from sampling site 6, and the following numbers indicate the isolation medium (1–16 as before) and the remaining digits refer to the number of the isolate from that source, as illustrated by KC712, isolate 12 from non-sporulating agar and; (c) if only two digits appear after “KC” (KC06, KC13, etc.), this indicates a pilot isolation study from the Karakum Desert and that the strains isolated from starch-casein agar from sampling site 6
Sixty-eight of the actinomycete isolates are considered to represent putative novel species, as their 16 S rRNA gene sequence similarities to the closest known type strains fall below the 99.3% threshold. This threshold was conservatively adopted in this study to indicate high novelty potential, particularly in genera such as Actinomadura, Micromonospora, Nocardioides, Nonomuraea, and Streptomyces (with 7, 9, 1, 27, and 15 isolates, respectively), which are widely distributed in desert habitats. In addition, there are putative novel species, two isolates from the genus Actinocorallia and one isolate each from the genera Agromyces, Kribbella, Microbacterium, Plantactinospora and Spongiactinospora that have rarely been isolated from desert biomes (Table S3). The relationships between putatively novel isolates and their closest phylogenetic relatives are shown (Figs. 6 and 7). 36 of the isolates (14%) belong to taxa rarely, if ever, isolated from arid desert habitats, namely the genera Actinocorallia (3 isolates), Agromyces (1), Jiangella (5), Kribbella (2), Microbacterium (1), Nocardiopsis (6), Plantactinospora (6), Saccharomonospora (1), Saccharopolyspora (10) and Spongiactinospora (1) (Figures S1-S10). Eleven of these isolates (31%) were separated from the type strains of their closest phylogenetic neighbours by relatively long branches and 16 S rRNA gene sequence similarities at or below the 99.3% threshold (Table S3). Eleven of the isolates shared identical or almost identical 16 S rRNA gene sequence similarities with the type strains of validly named species, as shown by relationships between isolates KC101, KC103 and KC408 and Saccharopolyspora erythraea, which produces the clinically important macrolide antibiotic, erythromycin A (Cortes et al. 1990). The remaining 223 isolates (86%) representing the colour-groups were assigned to genera known to be common in desert biomes, including Actinomadura (22 isolates), Micromonospora (37), Nocardia (10), Nocardioides (1), Nonomuraea (35), Pseudonocardia (12) and Streptomyces (106) (Figures S11 to S16). Fifty-seven of these isolates (26%) were considered to belong to putative novel species using the 99.3% cut-off point. On the other hand, fifty-six isolates shared identical or almost-identical 16 S rRNA gene sequence similarities with the type strains of their nearest phylogenetic neighbours. Identical sequences, for instance, were found between isolates 5K515, 6K523, and 7K401 and the type strain of Actinomadura geliboluensis.
Fig. 6. Abbreviated 16 S rRNA gene neighbor-joining trees showing relationships between isolates and their closest phylogenetic type strains in the genera Actinocorallia (a), Agromyces (b), Jiangella (c), Kribbella (d), Plantactinospora (e), Saccharopolyspora (f), Spongiactinospora (g), and Microbacterium (h). Scale bars are given in each of the figures. The phylogenetic trees were constructed based on 16 S rRNA gene sequence alignments of 1,371 bp (a), 1,395 bp (b), 1,440 bp (c), 1,334 bp (d), 1,370 bp (e), 1,270 bp (f), 1,316 bp (g), and 1,390 bp (h)
Fig. 7. Abbreviated 16 S rRNA gene neighbor-joining trees showing relationships between the isolates and their closest type strains in the genera Actinomadura (a), Micromonospora (b), Nocardioides (c), Nonomuraea (d), Pseudonocardia (e), and Streptomyces (f). Scale bars are given in each of the figures. The phylogenetic trees were constructed based on 16 S rRNA gene sequence alignments of 1,287 bp (a), 1,380 bp (b), 1,332 bp (c), 1,305 bp (d), 1,385 bp (e), and 1,301 bp (f)
Classification of isolates assigned to colour-groups
Most isolates assigned to colour-groups can be classified into genera and species given the distribution of representative marker strains included in the 16 S rRNA gene sequence analyses (Idris 2016; Kusuma 2020; Świecimska et al. 2023). In the present study, for instance, colour-group 88 encompasses all of the Actinocorallia strains whereas colour-groups 57 and 86 can be provisionally equated with two validly named species, A. geliboluensis and Actinomadura cremea (Table S4). Similarly, colour-groups 1, 20, 25, 72 and 85 can be equated with Micromonospora zamorensis, Micromonospora saelicesensis, Micromonospora avicenniae, Micromonospora citrea and Micromonospora fluostatini, and colour-groups 40, 42 and 76 with Nocardiopsis trehalosi, Nocardiopsis umidischolae and Nocardiopsis dassonvillei. It is also encouraging that the type strains of Kribbella turkmenica, Jiangella ureilytica, Nonomuraea diastatica, Nonomuraea mesophila, Saccharopolyspora terrae and Spongiactinospora gelatinilytica which formed distinct lineages in the phylogenomic tree (Fig. 8) were recovered as single-membered colour-groups (Table S3).
By far the most extensive taxonomic diversity was shown by isolates assigned to the genus Streptomyces (Table S4), including ones representing presumptive novel species (Table S2). Indeed, 41 out of the 88 multi-membered colour-groups (47%) were composed of streptomycetes, as exemplified by colour-group 31 which contains isolates shown to be phylogenetically close to the type strains of Streptomyces griseoincarnatus, Streptomyces leeuwenhoekii and Streptomyces thinghirensis. Another relatively large taxon, colour-group 50, consists of four isolates with identical 16 S rRNA gene sequences to the type strains of Streptomyces calvus, an isolate most closely related to Streptomyces albogriseolus and five similarly related to Streptomyces atrovirens.
A case in point is colour-group 23 which encompasses 10 isolates shown to be most closely related to the type strains of Micromonospora costi, Micromonospora halophytica, Micromonospora inositola and Micromonospora palomenae; seven of these isolates showed sequence similarities with their closest phylogenomic neighbours below the 99.3% threshold used in this study to delineate potentially new actinomycete species.
Ten of the remaining colour-groups (11.4%) encompassed isolates assigned to the genus Nonomuraea. These taxa included colour-group 67 which contains 5 isolates, including Nonomuraea deserti KC310^T^, that are most closely related to the type strains of Nonomuraea candida. Similarly, colour-group 82 consists of nine isolates, including Nonomuraea longispora KC201^T^, which also showed sequence similarities below the 99.3% threshold with the type strains of Nonomuraea salmonea. In addition, isolate 1K501 (colour group 44) and isolates 5K134 and 5K322 (colour-group 70) can be considered to be presumptive novel Nonomuraea species given their low sequence similarities (98.82 and 98.75%, respectively) with the type strains of Nonomuraea ceibae and Nonomuraea maheshkhaliensis, respectively.
In terms of colour-group numbers, less taxonomic diversity was found amongst isolates distributed to the remaining genera (Table S4). The number of colour-groups ranged from one with respect to isolates assigned to the genera Actinomadura, Agromyces, Kribbella and Saccharomonospora to five in the case of Actinomadura, as illustrated by colour-group 47 which contains isolates KC345 and 6K520 that are most closely related to the type strains of Actinomadura bangladeshensis and Actinomadura livida, respectively. Only three of the colour-groups included isolates belonging to more than one genus, notably colour-group 4, which included five isolates that shared high sequence similarities (99.44 to 99.51%) with Plantactinospora veratri NEAU-FHS4^T^, isolate 5K110, a presumptive novel Microbacterium species, and Micromonospora deserti 13K206^T^, which is most closely related (98.54% sequence similarity) to Micromonospora nigra DSM 43,818^T^.
Antimicrobial activity of selected isolates
The results obtained when rare and presumptive novel isolates were screened against the panel of bacterial and fungal strains (Table 6). Many of the isolates inhibited the growth of the P. aeruginosa (44%), C. albicans (26%), and S. aureus (25%) strains. In contrast, few isolates showed activity against the B. subtilis, E. faecalis and E. coli strains. It is particularly interesting that most of the Actinomadura isolates, several of the Micromonospora, Nonomuraea and Streptomyces strains, J. aurantiaca 8K307^T^, J. ureilytica KC603^T^, Sps. aridisoli 16K404^T^, Sps. elongata 7K502^T^, Sps. karakumensis 5K548^T^ and Sps. terrae 16K307^T^ inhibited the growth of P. aeruginosa ATCC 27,853, as did K. turkmeniaca 16K104^T^ and Microbacterium 5K110. Five out of the 8 isolates that inhibited the E. faecalis strain belonged to the genus Nonomuraea. Among the isolates, only two Streptomyces isolates, 13K109 and KC501, inhibited the growth of A. brasiliensis ATCC 16,404. None of the isolates showed activity against K. quasipneumoniae ATCC 700,603. Antimicrobial activity was not shown by isolates assigned to the genera Nocardioides, Plantactinospora, Pseudonocardia or Spongiactinospora. Overall, 32 out of the 57 isolates (56%) inhibited (≥ 10 mm) the growth of one or more of the reference strains.
Table 6. Inhibition zones (mm) shown by representative isolates from the Karakum desert in the agar diffusion assays after incubation for 24–48 h at optimal temperatures. In total, 57 isolates were analysed for their antimicrobial activity. Inhibition zones ≥ 10 mm were considered positive for antimicrobial activity. 1, Aspergillus Brasiliensis ATCC 16,404; 2, Bacillus subtilis ATCC 6633; 3, Candida albicans ATCC 10,231; 4, Enterococcus faecalis ATCC 29,212; 5, Escherichia coli ATCC 25,922; 6, Pseudomonas aeruginosa ATCC 27,853; 7, Staphylococcus aureus ATCC 25,923Strain1234567 Actinomadura 5K555--9--1855 ^#^6K520-----12- ^#^7K507--6--5- 7K515, 7K533, ^#^7K534------- 10K521-181489- ^#^KC06-2076- ^#^KC216--5--14- ^#^KC345----10- Jiangella *^,#^5K138^T^------- *^,#^8K307^T^-----14- *^,#^KC603^T^--6--7- Kribbella *^,#^16K104^T^--12--1220 Microbacterium *^,#^5K110-----9- Micromonospora ^#^13K206^T^----20- - ^#^15K316-----6- ^#^KC207------20 ^#^KC213-----12- ^#^KC606-----7- ^#^KC721, ^#^KC723------- Nocardioides ^#^KC13^T^------- Nonomuraea 1K501------- 4K202------7 5K322--10---- ^#^6K102^T^------- 7K110--8---- 7K523--7---13 8K306-----7- 15K203--8--11- ^#^KC201^T^---23-- - ^#^KC310^T^---22-- - ^#^KC333^T^-15-23-725 ^#^KC401---18-- - ^#^KC712^T^---24-- -
Plantactinospora *KC601, *KC728------- Pseudonocardia 5K418, KC304------- Saccharopolyspora *^,#^5K548^T^-----16- *^,#^7K502^T^---58-1553 *^,#^16K309^T^--12--1240 *^,#^16K404^T^-----12- Spongiactinospora ^,#^7K107^T^------- Streptomyces 3K401------- 5K209-----712 6K502--14--25- ^#^8K308---28--30 10K307------- 11K402--60---58 13K105--12---52 13K10928----12- ^#^13K301^T^------47 16K105-35----- 16K401------- KC50118-13--1848, representative of rare genera. ^#^, isolates selected for whole-genome sequencing, the remaining strains represent putatively novel species
Genomic features of selected isolates
The genome features of the 32 isolates used to generate the phylogenomic tree are shown (Table 7). The sizes of their draft genomes of the strains ranged from 3.71 Mbp for Microbacterium 5K110 to 10.86 Mbp for Non. diastatica KC712^T^. Many of the isolates can be considered to be extremely gifted sensu Baltz (2017) as they have large genomes (≥ 8 Mbp) and hence can be expected to be rich in BGCs encoding for known and novel specialized metabolites. Similarly, the remaining isolates, apart from the Microbacterium strain and Sps. elongata 7K502^T^, are moderately gifted with genome sizes ranging from 5.30 to 7.76 Mbp in Noc. turkmenicus KC13^T^ and Streptomyces 8K308, respectively. The digital (d) G + C contents of the isolates ranged from 69.4% in K. turkmenica 16K104^T^ to 72.5% in J. ureilytica KC603^T^ and Streptomyces 8K308, respectively. The lowest number of protein coding genes, 3,466, was recorded for the Kribbella strain and the highest number, 9,740 for Non. diastatica KC712^T^. The number of rRNA and tRNA operons found in the genomes of the isolates varied from 3 to 17 and from 44 to 79.
The genomes sizes of the Actinomadura isolates (8.09–9.80 Mbp) are well within the range (6.17–10.26 Mbp) reported for type strains of Actinomadura species. Similar deductions can be drawn from analyses of the Kribbella, Nonomuraea, Spongiactinospora, Streptomyces, Jiangella and Micromonospora isolates. In contrast, the genome sizes of the type strains of Saccharopolyspora, namely 6.29–9.02 Mbp (Nouioui et al. 2019), were exceeded by that of Sps. elongata 7K502^T^ with a genome size of 10.30 Mbp. It is also interesting that Actinomadura isolates 6K520 and 7K534 have similar genomic features as they formed a distinct lineage in the phylogenomic tree (Fig. 8). The genome of Noc. turkmenicus KC13^T^ (5.3 Mbp) is larger than expected for a Nocardioides strain, but its dDNA G + C content is within the normal range for members of the genus Nocardioides. Similarly, Microbacterium isolate 5K110, like other Microbacterium strains, has a relatively large genome (3.7 Mbp) and a high dDNA G + C content (70.0). These values place the isolate at or near the top of the ranges shown by Microbacterium type strains.
Table 7. Genome features of presumptively novel actinomycetes. 32 isolates underwent whole-genome sequencing and genomic analysesGenera and strainsGenomeDigital DNA G + C content (mol%)ContigsGenesProtein coding genesrRNA operonstRNA operonsGenBank accession numbersrange (in bp)coverage Actinomadura 6K5208,190,264159x71.94457,5807,2901161SMLC00000000 7K5079,799,14445x71.35639,0538,6981463SMKK00000000 7K5348,085,395172x72.15657,5397,2441461SMKB00000000 KC069,380,42774x71.34218,6828,328967SMKT00000000 KC2169,063,53231x71.36678,5768,2151179SMJX00000000 KC3458,265,01034x71.86007,9697,618763SMKH00000000 Jiangella 5K138^T^7,034,06388x71.01346,4076,207345SMKZ00000000 8K307^T^6,620,775152x71.81026,0105,816544SMLB00000000 KC603^T^6,345,964105x72.51685,8345,632545SMKL00000000 Kribbella 16K104^T^7,447,14220x69.43517,1656,877345SMKR00000000 Microbacterium 5K1103,713,48838x70.01793,5933,466448VBUM00000000 Micromonospora 13K206^T^6,720,28730x72.46156,4155,981649POUB00000000 15K3166,957,401138x72.44556,4576,147559SMKG00000000 KC2077,188,14631x72.26376,5886,015350SMKJ00000000 KC2136,426,73365x71.63636,1315,772753SMKF00000000 KC6066,658,79843x71.74296,3085,833748SMKN00000000 KC7216,461,179118x71.64416,1615,780779SMJY00000000 KC7236,107,06884x72.22925,7405,350766SMKD00000000 Nocardioides KC13^T^5,300,16263x69.7475,1064,962647JAALAA000000000 Nonomuraea 6K102^T^10,138,47897x70.84279,6829,2171262SMLD00000000 KC201^T^9,164,34147x70.64658,7898,2811266SMJZ00000000 KC310^T^10,694,046144x70.947010,1459,541970SMKO00000000 KC333^T^9,868,67230x71.37299,7719,2791765POUD00000000 KC4019,792,88273x70.64949,4228,943969VBUN00000000 KC712^T^10,861,99370x70.348810,4599,740566SMKP00000000 Saccharopolyspora 5K548^T^6,670,71376x69.71376,2345,983550SMLA00000000 7K502^T^10,298,286250x69.72779,3268,7511151SMKW00000000 16K309^T^6,653,094112x69.71676,2436,004751SMKS00000000 16K404^T^6,008,184163x69.6835,3895,147851SMKV00000000 Spongiactinospora 7K107^T^8,049,10730x70.89578,0337,513861POUA00000000 Streptomyces 8K3087,755,21789x72.57037,3416,676966SMKC00000000 13K301^T^9,035,42130x70.810248,8808,1381569POUC00000000
Phylogenomic relationships of taxonomically diverse isolates
The assignment of the selected isolates to well supported clades corresponding to the genera Actinomadura, Jiangella, Micromonospora, Nonomuraea, Saccharopolyspora and Streptomyces in the phylogenomic tree (Fig. 8a and b) confirm the generic relationships found in the corresponding 16 S rRNA gene trees (Figs. 6 and 7). It is also evident that several of the isolates belong to well supported lineages which correspond to ones found in the phylogenetic trees, as exemplified by the assignment of Actinomadura isolates KC345 and 7K507, 6K520 and KC06 and KC216 to distinct, albeit related subclades (Fig. 8). The phylogenomic analysis also highlights presumptive novel isolates, including Noc. turkmenicus KC13^T^ and Microbacterium 5K110, as well as ones which correspond to known lineages, as exemplified by J. asiatica 5K318^T^, Non. deserti KC310^T^ and Sps. elongata 7K502^T^.
Fig. 8. Phylogenomic tree showing relationships between isolates belonging to the genera (a) Actinomadura, Kribbella, Microbacterium, Nocardioides, Nonomuraea and Streptomyces, and (b) Spongiactinospora, Jiangella, Micromonospora and Saccharopolyspora. The numbers above branches are GBDP pseudo-bootstrap support values > 60% from 100 replications, with average branch supports of 96.1% and 94.2%, respectively. Trees were inferred with FastME 2.1.6.1 (Lefort et al. 2015) from GBDP distances calculated from genome sequences. The branch lengths are scaled in terms of the GBDP distance formula d_5_. The trees were rooted at the midpoint (Farris 1972). δ statistics were 0.103 and 0.12, respectively
Biosynthetic gene clusters
Several of the BGCs detected in the genomes of the Actinomadura isolates showed more than 50% gene identity with known bioclusters and were unique to these strains. For example, isolates KC06 and 7K507 harbored bioclusters partially similar to the macrocyclic lactams ML-449 (62% gene identity) (Jørgensen et al. 2010) and vicenistatin (55% gene identity) (Shindo et al. 1993), respectively. Similarly, the genome of Actinomadura 7K534 contained BGCs related to the biosynthesis of napyradiomycin (53% gene identity), an antioxidant with anti-inflammatory activity (Choi et al. 2020), and 7-prenylisatin (60% gene identity), an isatin-type antibiotic active against Bacillus subtilis (Liang and Wang 2019). Additionally, isolates KC06 and KC216 possessed BGCs similar to those associated with catenulipeptin (60% gene identity), a class III lanthipeptide (Wang and van der Donk 2012), and citrulassin D (80% and 100% gene identity, respectively), a lasso peptide (Tietz et al. 2017).
The genomes of the Nonomuraea isolates, including the type strains of Non. deserti, Non. diastatica, Non. longispora, and Non. mesophila (Saygin et al. 2020d), harbored biosynthetic gene clusters (BGCs) showing 100% gene identity with clusters responsible for the production of alkylresorcinol, a common phenolic lipid (Kozubek and Tyman 2005), and rhizomide A, a compound exhibiting weak antitumor activity (Wang et al. 2018). Furthermore, these genomes also contained BGCs known to encode indigoidine (80% gene identity), a natural pigment with antioxidant and antimicrobial properties (Yumusak et al. 2019), and GE2270 (80% gene identity), an antibiotic that inhibits protein synthesis in bacteria (Kettenring et al. 1991). In addition, all of the Nonomuraea isolates harbored BGCs associated with the production of chlortetracycline (5% gene identity), a broad-spectrum antibiotic active against a range of bacteria (Pulicharla et al. 2015).
As in earlier studies (Carro et al. 2018, 2019a), many of the BGCs detected in the genomes of the Micromonospora isolates were strain-specific. For instance, isolates KC207, KC213, KC606, KC721, KC723, 13K206, and 15K316 harbored bioclusters partially similar to known compounds. These included tirandamycin (40% gene identity), an antibiotic that inhibits transcription by disrupting the bacterial RNA polymerase (Reusser 1976); thiocoraline (5% gene identity), a depsipeptide exhibiting antitumoral activity (Sparidans et al. 1999); tallysomycin A (5% gene identity), a glycopeptide-derived antitumor antibiotic (Galm et al. 2011); colabomycin E (9% gene identity), an anti-inflammatory agent that inhibits caspase 1 and the synthesis of interleukins (Petříčková et al. 2014); hedamycin (12% gene identity), a polyketide antibiotic with anticancer properties (Hansen et al. 1995); azicemicin B (13% gene identity), an antimicrobial compound active against Gram-positive bacteria, including mycobacteria (Tsuchida et al. 1995); and crochelin A (11% gene identity), an unusual siderophore (Baars et al. 2018).
The genomes of the Saccharopolyspora strains were especially rich in BGCs predicted to encode a broad range of novel antibiotics. Specifically, Sps. aridisoli 16K404^T^, Sps. elongata 7K502^T^, Sps. karakumensis 5K548^T^, and Sps. terrae 16K309^T^ contained BGCs associated with the production of lankacidin C (13–20% gene identity), an antibiotic with antitumor activity (Ayoub et al. 2019), and the siderophore staphylobactin (25% gene identity). Additionally, Sps. aridisoli 16K404^T^, Sps. karakumensis 5K548^T^, and Sps. terrae 16K309^T^ harbored gene clusters predicted to encode a specialized metabolite similar to saquayamycin A, an aquayamycin antibiotic active against Gram-positive bacteria (Uchida et al. 1985). Furthermore, Sps. karakumensis 5K548^T^ and Sps. terrae 16K309^T^ possessed genes associated with the biosynthesis of pepticinnamin E (6% gene identity), a farnesyl-transferase inhibitor with potential applications in cancer and malaria treatment (Santa Maria et al. 2019), and brasilicardin A (30% gene identity), a diterpenoid antibiotic with immunosuppressive properties (Komaki et al. 1999).
The genome of Spongiactinospora gelatinilytica 7K107^T^ contained BGCs associated with a variety of bioactive compounds. These included amipurimycin (23% gene identity), a nucleoside antibiotic with antimicrobial activity (Harada and Kishi 1977; Iwasa et al. 1977); epoxomicin (25% gene identity), a proteasome inhibitor with in vivo anti-inflammatory properties (Meng et al. 1999); kedarcidin (5% gene identity), a chromoprotein antitumor antibiotic (Leet et al. 1992); and lobosamides A, B, and C (21% gene identity), polyene macrolactams active against the protozoan parasite Trypanosoma brucei (Schulze et al. 2015).
Stm. cahuitamycinicus 13K301^T^ and Streptomyces 8K308 harbored BGCs similar to those encoding the siderophore desferrioxamine E (50% and 83% gene identity, respectively) and ficellomycin (3% gene identity in both isolates), an aziridine antibiotic with potential therapeutic value (Liu et al. 2017; He et al. 2018). However, only isolate 8K308 contained BGCs associated with the synthesis of azalomycin F3a (26% gene identity), a macrocyclic polyketide (Zhai et al. 2020); herboxidiene (2% gene identity), a polyketide exhibiting herbicidal activity (Miller-Wideman et al. 1992); pentostatin (6% gene identity), a purine nucleoside antibiotic used to treat hematological cancers (Wu et al. 2017); and spiramycin (8% gene identity), a macrolide antibiotic used to treat Toxoplasma gondii infections during pregnancy (Montoya et al. 2021). In contrast, only Stm. cahuitamycinicus 13K301^T^ was found to harbor BGCs associated with the production of albaflavenone (100% gene identity), a sesquiterpene antibiotic exhibiting antibacterial activity (Zhao et al. 2008), and a bioactive metabolite similar to neocarzinostatin (41% gene identity), a chromoprotein antitumor antibiotic (Edo and Koide 1997).
Although all of the Jiangella strains were found to have the capacity to synthesize alkylresorcinol (100% gene identity), most of the BGCs were strain-specific. The genome of J. aurantiaca 8K307^T^ contained BGCs associated with the production of tetarimycin A (5% gene identity), a compound that inhibits methicillin-resistant Staphylococcus aureus strains (Kallifidas et al. 2012), whereas the genome of J. asiatica 5K138^T^ harbored BGCs related to the production of acumicin (4% gene identity), a cyclic depsipeptide with anti-mycobacterial activity (Hawkins et al. 2018). Finally, the genome of J. ureilytica KC603^T^ contained BGCs partially similar to those encoding primycin (5% gene identity), an antibiotic active against Gram-positive bacteria (Horváth et al. 1979), and LL-D49194α1 (7% gene identity), an antitumor antibiotic (Dong et al. 2019).
Four out of the ten BGCs detected in the genome of K. turkmenica 16K104^T^ were associated with the production of alkylresorcinol (100% gene identity), catenulipeptin (60% gene identity), and ficellomycin (9% gene identity), which were mentioned previously. Five out of the six BGCs identified in the genome of Microbacterium 5K110 were associated with the synthesis of known compounds, including carotenoid pigment (50% gene identity) and berninamycin A (26% gene identity), a cyclic thiopeptide antibiotic that inhibits protein synthesis in Gram-positive bacteria (Thompson et al. 1982). The remaining BGC is predicted to encode a novel metabolite. The genome of Noc. turkmenicus KC13^T^ contained six BGCs, three of which are novel. The remaining BGCs were associated with the production of desferrioxamine B (60% gene identity), a siderophore used to remove excess iron in patients with transfusion-related hemoglobin disorders (Codd et al. 2018); dynemicin A (5% gene identity), an anthraquinone antibiotic that inhibits Gram-positive bacteria (Konishi et al. 1989); and kijanimicin (4% gene identity), a macrolide antibiotic with antibacterial and antitumor properties (Waitz et al. 1981; Bradner et al. 1983).
Plant growth promoting properties of selected isolates
The genomes of the isolates, notably those of the Jiangella strains, were rich in genes associated with phosphate metabolism and regulation (Table S5), as exemplified by genes ppa, phoR, pstS and phoU which express for solubilisation of inorganic phosphate (Lahti et al. 1988), a phosphate transport regulatory protein (Lamarche et al. 2008), a periplasmic protein involved in the synthesis of the phosphate ABC transporter (Willsky and Malamy 1980), and a transport protein regulating metabolism (Muda et al. 1992), respectively. In addition, all of the jiangellae have the genetic capacity to produce a range of phosphatases. In contrast, the genomes of none of the isolates contained genes associated with nitrogen fixation. Apart from the Spongiactinospora strain, the genomes of all of the isolates contain genes predicted to encode for indole − 3- glycerol phosphate synthase, the precursor of IAA in the tryptophan biosynthetic pathway in plants (Ouyang et al. 2000). They also have genes encoding for other components of this pathway, including aminase (trpA and trpB), anthranilate synthase (trpE) and anthranilate phosphoribosyl transferase (trpD) (Lambrecht and Downs 2013). The level of expression of IAA is influenced by the biosynthetic pathway, the location of the genes involved and their regulation (Duca et al. 2014). The genomes of most of the isolates were equipped with genes associated with the synthesis of chitinases, endoxylanase and ß-glucosidases. In contrast, the production of pectate lyase and 1,4-β-xylosidase was mainly restricted to the Jiangella, Micromonospora, Nonomuraea and Streptomyces strains. Genes predicted to encode for cutinases, which hydrolyse cutin ester bonds (Chen et al. 2013), were detected in the genomes of Micromonospora isolates KC213 and KC721, and Stm. cahuitamycinicus 13K301^T^. Cellulases (Juturu and Wu 2014), chitinases (Oyeleye and Normi 2018) and pectinases (Liu and Kokare 2017) are also noted for their industrial potential.
Genes predicted to encode for siderophores are discontinuously distributed across the genomes of the isolates (Table S6). In contrast, the genetic capacity of the isolates to produce ferric iron ABC transporters is restricted to the genera Actinomadura, Jiangella, Kribbella and Micromonospora whereas only Micromonospora isolates KC723 and 15K316, and Stm. cahuitamycinicus 13K301^T^ have more genes associated with the synthesis of desferrioxamine E (Smits and Duffy 2011).
Genes of representative isolates associated with adaptation to desert habitats
The genomes of all of the isolates contained genes (e.g. dnaK, grpE, hrcA, groL and groES) linked with heat shock responses (Li et al. 2011) though only Non. aridisoli KC333^T^ contained genes cspA and cspC, which express for protein families that respond to cold shock (Jones and Inouye 1994). In contrast, without exception, the isolates had the capacity to produce cold shock protein SCO 4325. Other common genes include ahpD, which encodes for the synthesis of alkyl hydroperoxide reductase, an enzyme involved in compensatory interactions amongst hydrogen peroxide stress genes (Bsat et al. 1996) and in the protection of DNA against oxidative damage (Jacobson et al. 1989). Several isolates, especially those assigned to the genera Kribbella, Nocardioides, Nonomuraea, Saccharopolyspora and Streptomyces, have the ability to synthesize sarcosine oxidase, which is also associated with oxidative stress responses (Kappes et al. 1999; Mandon et al. 2003). Similarly, all of the strains, apart from the Jiangella and Spongiactinospora isolates, contained katG, a gene that encodes for catalase peroxidase, an enzyme which offers protection against reactive oxygen species (Normand et al. 2012). Many of the isolates, particularly the Jiangella and Micromonospora strains, have the potential to produce aquaporin Z, a protein that mediates the transport of water molecules across cell membranes (Johansson et al. 2000). Genes associated with carotenoid biosynthesis (e.g. carB, crtB and crtL) were found mainly in the genomes of the Actinomadura and Saccharopolyspora isolates (Table S7). Genes encoding for choline dehydrogenase (chdH) and choline kinase (chkA) were restricted mainly to the genomes of the Nonomuraea isolates whereas gene betC, which expresses for choline sulfatase, was detected in the genome of the Jiangella and Micromonospora strains, these genes are associated with oxidative stress and uptake of betaine and choline (Boncompagni et al. 1999, 2000; Nau-Wagner et al. 2012). The genomes of all of the isolates contained genes predicted to encode for copper resistance, as exemplified by copC and copD, which mediate resistance to copper by accumulating it in the periplasm and membrane proteins (Cha and Cooksey 1991). These genes were detected in all of the isolates, apart from Actinomadura KC06 and J. asiatica 5K138^T^ in the case of the former and Spa. gelatinilytica 7K107^T^ with respect to the latter. In contrast, the capacity to resist arsenic was restricted to Microbacterium 5K110, Micromonospora KC207 and 15K316, Noc. turkmenicus KC13^T^, Non. diastatica KC201^T^, Non. longispora KC201^T^, Sps. elongata 7K502^T^ and Sps. terrae 16K309^T^. Interestingly, resistance to chromium compounds was limited to the five Nonomuraea strains, Microbacterium 5K110 and Micromonospora KC207 and Noc. turkmenicus KC13^T^. The genomes of all isolates encode DNA gyrase subunits A and B, which are essential for DNA replication and supercoiling. Although these enzymes are not inherently involved in antibiotic resistance, mutations in their encoding genes (gyrA and gyrB) have been associated with resistance to fluoroquinolones (Ruiz 2003). Similarly, most of the isolates have the capacity to produce β-lactamases. In contrast, resistance to vancomycin is found to be rare among the isolates.
The genomes of all of the isolates contained genes associated with DNA repair (Table S8), as exemplified by the UvrABC excinuclease system which repairs DNA damage by cutting DNA strands on both sides of the damaged region (Sancar and Rupp 1983). Similarly, all of the isolates harboured genes predicted to express for ATP-dependent DNA ligases (ligC and ligD), DNA repair proteins (radA and recN), exodeoxyribonucleases (III, VII small and large subunits), endonucleases (III, IV and VIII), RecA protein and SOS-response repressor and protease (lexA). Three genes, recF, recO and recR, which are associated with DNA recombination and repair proteins, were detected in all the genomes, except K. turkmeniaca 16K104^T^. This isolate was one of the few strains with the capacity to produce exodeoxyribonuclease V alpha (recD), beta (recB) and gamma (recC) chains, which are part of the bacterial RecBCD pathway (Pavankumar et al. 2010).
The genomes of all of the isolates contained whiB, a gene which encodes sporulation protein WhiB that is needed for the differentiation of aerial hyphae into mature spores in Streptomyces coelicolor (Molle et al. 2000). Gene whiD, which also contributes to spore differentiation, was detected in the genomes of the Jiangella, Micromonospora, Saccharopolyspora and Streptomyces strains. Several of the isolates contained genes (e.g. whiE-ks, whiE-clf, whiE VII) that express for spore pigments in Streptomyces strains (Swiercz and Elliot 2012), as illustrated by whiE VII, which is present in the genomes of all of the Micromonospora strains, most of the Actinomadura and Nonomuraea isolates, and in Spa. gelatinilytica 7K107^T^ and Stm. cahuitamycinicus 13K301^T^ (Table S9).
Discussion
Since the assignment of filamentous actinomycetes, notably streptomycetes, to colour-groups provide an insight into the level of cultivable actinomycete diversity in natural habitats (Goodfellow et al. 2018; Świecimska et al. 2023), it can be concluded that the Karakum Desert habitats contain diverse communities of mainly filamentous actinomycetes. The extent of this diversity is similar to that found in corresponding studies on hyper-arid and extremely hyper-arid Atacama Desert soils (Okoro et al. 2009; Busarakam 2014; Idris 2016; Świecimska et al. 2023). The isolates obtained from this study were assigned to 17 genera, 11 families and 8 orders (Table 5). A similarly, broad range of taxa were found in comparable surveys of actinomycete communities in Atacama Desert soils (Busarakam 2014; Idris 2016; Goodfellow et al. 2018) though representatives of the families Jiangellaceae and Microbacteriaceae were not detected in these studies. In contrast, putative novel Actinomadura, Kribbella, Nocardia, Nocardiopsis, Nonomuraea, Pseudonocardia and Streptomyces strains were isolated from both Atacama and Karakum Desert soils. Streptomycetes isolated from these biomes and from the Kazakhstan Desert (Ziyat et al. 2019) proved to be the richest source of presumptive novel species. An interesting feature of the present study was the isolation of presumptive novel members of rare genera such as Jiangella, Plantactinospora and Spongiactinospora. Several of the selective media did not to support the growth of the target actinomycetes but did allow that of non-target taxa. The failure, for instance, to isolate Amycolatopsis strains on SM1, SM2 and SM3 agar media designed for this purpose (Tan et al. 2006) may be due to their absence in the Karakum Desert environmental samples whereas the growth of members of other taxa, such as Actinocorallia, Actinomadura, Micromonospora, Nocardia, Pseudonocardia, Saccharopolyspora and Streptomyces, can be attributed to the lack of competition on the isolation plates reflecting the low incidence of actinomycete propagules in the soil suspensions used to inoculate them. Similar “anomalous” results have been reported previously (Vickers et al. 1984; Busarakam 2014).
AntiSMASH predicts BGCs and potential natural products based on the percentages of genes from the closest known bioclusters which show BLAST hits with corresponding BGCs in the genomes under consideration. The number of bioclusters found in the genomes of the strains ranges from 4 in the genome of Microbacterium 5K110 to 50 in that of Micromonospora KC207. The genomes of all of the isolates contained bioclusters predicted to encode for core metabolites like ectoine, a protective molecule which enables bacteria to counter extreme conditions (Graf et al. 2008), arylpolyene-like compounds that are structurally and functionally similar to carotenoids (Schöner et al. 2016) and which show antimicrobial and antioxidant activity (Narsing Rao et al. 2017), geosmin, a volatile terpene responsible for earthy odours (Gerber and Lechevalier 1965) and melanin pigments which have antioxidant, antitumor and antimicrobial activity and protect microorganisms from UV-radiation (Vasanthabharathi and Jayalakshmi 2020). In contrast, most of the bioclusters predicted to encode for druggable molecules, including antibiotics, were discontinuously distributed across the genomes of the isolates. In general, the genomes of the selected isolates showed similar patterns of BGCs, notably ones predicted to encode for non-ribosomal peptide synthetases (NRPS), type I polyketide synthases (T1PKS), lanthipeptides and terpenes. Microorganisms which contain lanthipeptide gene clusters tend produce novel compounds, such as antibiotics, which inhibit the growth of multidrug-resistant S. aureus (Repka et al. 2017). The average number of NRPS bioclusters per genome was 4 out of 135 bioclusters, the corresponding numbers for the T1PKS and terpenes were 3 (112 bioclusters) and 4 (140 bioclusters), respectively. Many of the bioclusters were discontinuously distributed across the phylogenomic lineages with some shown to be taxon and strain specific, results that are in agreement with recent data on closely related Streptomyces species (Vicente et al. 2018; Park and Andam 2019; Martinet et al. 2020). Critically, 336 of the 861 BGCs (39%) did not show any similarity to known compounds while the number of bioclusters sharing more than 50% gene identity with known metabolites was low at 130 bioclusters (15%). Actinomadura and Nonomuraea strains are difficult to recognise on isolation plates and because of this they have rarely featured in bioprospecting campaigns even though it is apparent that they have the ability to produce new bioactive compounds (Sungthong and Nakaew 2015; Saygin et al. 2020d; Ay 2021). Eighty out of the 216 bioclusters (37%) detected in the genomes of the Actinomadura strains are associated with the production of new specialized metabolites. Similarly, 97 from a total of 204 bioclusters (48%) found in the genomes of the Nonomuraea strains were predicted to encode for new, uncharacterized compounds. Micromonospora and Saccharopolyspora strains are important sources of new antibiotics, especially ones of therapeutic value (Bérdy 2005; Hifnawy et al. 2020; Sayed et al. 2020b). However, their full potential in this respect only become apparent when it was shown that they had large genomes predicted to encode for new and uncharacterized specialized metabolites (Carro et al. 2018, 2019a, b). In this study, the genomes of the Micromonospora isolates, including M. deserti 13K206^T^ (Saygin et al. 2020c), contained between 15 and 49 BGCs, notably ones associated with the production of antibiotics, siderophores and terpenes, results similar to those reported by Carro et al. (2018). The genomes of Sps. aridisoli 16K404^T^, Sps. elongata 7K502^T^, Sps. karakumensis 5K548^T^ and Sps. terrae 16K309^T^ harbored between 18 and 29 bioclusters predicted to encoded for a broad range of specialized compounds, particularly antibiotics. Sixty one out of the 171 bioclusters detected in the genomes of the micromonosporae (36%) are associated with the synthesis of novel antibiotics; the corresponding numbers for the Saccharopolyspora strains; are 33 and 100 BGCs (33%), respectively. Isolate 7K107^T^, a Karakum Desert strain, was proposed as the type strain of Desertactinospora gelatinilytica (Saygin et al. 2019b) then transferred to the genus Spongiactinospora as Spa. gelatinilytica (Ay et al. 2020). The type strain of Spa. gelatinilytica has a large genome rich in BGCs. It is particularly interesting that 16 out of the 38 bioclusters detected in the genome of isolate 7K107^T^ did not show any similarity to known BGCs. The Streptomyces isolates, Stm. cahuitamycinicus 13K301^T^ and Streptomyces 8K308, have large genomes replete in BGCs. The genomes of these strains contained 34 and 29 bioclusters, 13 (38,2%) and 15 (51,7%) of which did not show any similarity to ones coding for known compounds. As expected, the genome of the moderately gifted Jiangella, Kribbella, Microbacterium and Nocardioides isolates have fewer BGCs than their extremely gifted counterparts. Members of these taxa rarely feature in bioprospecting campaigns although they are known to produce bioactive compounds (Gullo et al. 1988; Matson and Bush 1989; Matson et al. 1993; Alitalo et al. 2003; Li et al. 2004; El-Refai et al. 2011; Han et al. 2014; Jiao et al. 2017). In this study, 11 out of the 21 BGCs detected in the genomes of the Jiangella isolates did not show any similarity to known bioclusters thereby providing further evidence that members of this genus are a potential source of new antibiotics. Six out of the ten BGCs detected in the genome of K. turkmenica 16K104^T^ were novel whereas Noc. turkmenicus KC13^T^ contains six BGCs, 3 of which are novel, and a novel BGC in Microbacterium 5K110.
Enhanced crop production and control of plant pathogens are key factors in reducing threats to global food security (Boukhatem et al. 2022). Free-living and mutualistic bacteria use direct and indirect mechanisms to promote and protect plant growth (Hayat et al. 2010; Glick 2012; Nouioui et al. 2019). Comparative mining of the genomes of the isolates revealed the presence of genes associated with direct (phosphate solubilisation, phytohormone production) and indirect (synthesis of lytic enzymes and siderophores) mechanisms beneficial to the growth and protection of plants. Bacteria and fungi synthesize siderophores in response to iron limitation (Saha et al. 2016). These compounds chelate ferric iron (Fe^+ 3^) thereby making iron available for microbial and plant cells (Kloepper et al. 1980). Taxonomically diverse rhizobacteria enhance the growth of plants by making iron available to them through the synthesis of siderophores (Ahmed and Holmström 2014; Grobelak and Hiller 2017) whereas the ability of the latter to bind iron tightly is used to control phytopathogens by restricting its availability (Köhl et al. 2019). Karakum Desert isolates are quite capable of promoting and protecting plant growth properties (Tables S5 and S6). Isolates belonging to the genus Micromonospora have a genome rich in genes associated with siderophore production compared to other genera.
Bacteria in extreme habitats have developed ways of responding to environmental stress (Abdel-Mageed et al. 2020). In the present study, the genomes of the isolates were found to contain between 18 and 44 putative stress-related genes, notably ones associated with carbon starvation, drought tolerance, oxidative and osmotic stress, detoxification and temperature fluxes. Some genes associated with environmental adaptation are common to all of the isolates but others are either discontinuously distributed or strain specific (Table S7). Similar results have been reported for actinomycetes isolated from hyper-arid Atacama Desert soils (Busarakam et al. 2016). Under nutrient-limiting conditions, such as those in desert soils, bacteria need to acquire and store carbon. The genomes of most of the Actinomadura strains and all of the Jiangella, Kribbella, Nocardioides, Nonomuraea, Saccharopolyspora and Streptomyces isolates contained a gene (cstA) that encodes for starvation protein A, which activates peptide uptake thereby indicating that strains are adapted to low carbon conditions (Rasmussen et al. 2013). It is also notable that the genomes of the Actinomadura, Nocardioides, Nonomuraea and Saccharopolyspora isolates, J. aurantiaca 8K307^T^ and Stm. cahuitamycinicus 13K301^T^ contain coxD and coxE genes, which encode CO uptake thereby showing that these strains have the ability to function as chemolithotrophs (Lorite et al. 2000), as is the case with Blastococcus, Geodermatophilus and Modestobacter strains that are abundant in the Atacama Desert soils (Idris et al. 2017; Bull et al. 2018; Golińska et al. 2020).
Conclusion
Novel, rare and gifted filamentous actinomycetes isolated from natural habitats, notably the extremebiosphere, remain a unique source of specialized metabolites of biotechnological importance, including antibiotics of clinical value and plant growth promoting compounds relevant to sustainable agriculture. Yet, the initial steps in natural product pipelines tend to be overshadowed by later stages of the process, such as chemical exploration of drug leads. Despite this, the selective isolation, dereplication, taxonomic characterization and screening of representative actinomycetes from neglected and unexplored extreme biomes provides a practical and effective way of selecting rare and novel isolates in the search for new bioactive compounds using state-of-the-art technologies, notably genome mining (Świecimska et al. 2023). In addition, advanced analytical procedures are now available to detect and dereplicate new specialized metabolites from complex biological extracts of selected isolates (Atanasov et al. 2021; Ossai et al. 2022) and marine sediments (Tuttle et al. 2019).
In the present study, the relevance of the early stages of natural product pipelines was revealed by the assignment of representative isolates from the Karakum Desert sampling sites to 17 genera, 11 families and eight orders of the phylum Actinomycetota. This extensive cultivable biodiversity included many novel and putatively new species belonging to rare taxa, such as members of the genera Actinocorallia, Actinomadura, Jiangella, Nonomuraea and Spongiactinospora, which encompasses strains which can be difficult to isolate from natural habitats. The isolation of members of such taxonomically diverse taxa reflects the use of a broad-range of selective media, as exemplified by the growth of novel and putatively novel Nonomuraea strains on SM1, SM3, marine, humic acid-vitamin, M1, non-sporulating, R2A, starch-casein and glycerol-asparagine agar. In contrast, isolates representing several taxa were isolated from plates of humic acid-vitamin which is known to be a relatively non-selective medium (Busarakam 2014; Idris 2016). This wealth of taxonomic data show that pristine Karakum Desert habitats are hot spots of filamentous actinomycetes that belong to taxa known to be a source of a chemically diverse specialized metabolites.
The results of the antimicrobial diffusion assays are particularly encouraging as nearly half of the representative isolates inhibited the growth of one or more of the reference strains. In general, these data are in line with those from corresponding results recorded for taxonomically diverse, filamentous actinomycetes isolated from hyper-arid and extreme hyper-arid Atacama Desert soils. The combined datasets shows that high hit rates can be achieved when dereplicated filamentous actinomycetes from arid desert habitats are examined in primary antimicrobial screens. Given the urgent need to find new antibiotics to control the spread of Gram-negative pathogens, it is particularly significant that Enterococcus faecalis ATCC 29,212 was inhibited by the Actinomadura, Nonomuraea, Saccharopolyspora and Streptomyces isolates. Also, Actinomadura, Jiangella and Saccharopolyspora isolates inhibited the growth of Pseudomonas aeruginosa ATCC 27,853, albeit less to.
The genomes of most of the novel and presumptive novel isolates representing ten of the genera were rich both in BGCs predicted to encode for an amazing array of specialized metabolites and genes associated with the production of compounds that promote and protect plant growth. Many of the BGCs were strain or taxon specific providing further evidence of the value of taxonomic approaches to drug discovery. Interestingly, the genomes of some of the isolates, such as the Jiangella, Micromonospora, Nonomuraea and Saccharopolyspora strains, contained bioclusters which did not show any similarity to ones expressing for known compounds thereby underscoring the significant of genome mining in the search for novel specialized metabolites. In contrast, the genomes of most of the strains, notably the Jiangella isolates, contained genes associated with phosphate metabolism whereas those predicted to encode for siderophores were discontinuously distributed across the genomes of the tested isolates.
It is not surprising that most extensive taxonomic diversity was shown by the isolates assigned to the genus Streptomyces as representatives of this taxon tend to be dominant in arid desert habitats. However, a unique feature of this culture-based study is that the representative streptomycetes were shown to belong to either putatively novel or rare Streptomyces species known or predicted to encode for new specialized metabolites. Streptomyces asenjonii and Streptomyces leeuwenhoekii, which were isolated from Atacama Desert, have capacity to produce new bioactive compounds with antibacterial, anti-cancer and anti-inflammatory properties. In this content, Stm. cahuitamycinicus 13K301^T^ and Streptomyces isolate 8K308 are of particular interest as most of the BGCs embedded in their genomes showed little, if any, similarity to ones coding for known compounds. These are significant developments as streptomycetes isolated from natural habitats remain the most important source of new chemically diverse antibiotics. Similar deductions can be drawn for the putatively novel and rare species that were shown to belong to the other genera, notably Micromonospora, Nonomuraea and Saccharopolyspora. Consequently, it can be concluded that the most tangible outcome of this culture-based bioprospecting strategy is the generation of a high quality library of novel and rare filamentous actinomycetes with the capacity to produce a multiplicity of novel specialized metabolites that can be exploited for useful purposes, such as drug discovery and the promotion and protection of plant growth. Further, work is required to build upon these developments not least the need to understand the ecological and evolutionary processes that drive the taxonomic and metabolic diversity of filamentous actinomycetes in Karakum Desert habitats.
It is encouraging that integrated strategies are being developed to improve natural product pipelines in order to promote the sustainable discovery and development of novel antibiotics (Miethke et al. 2021), and to foster an improved understanding of plant-microbe interactions to preserve natural ecosystems and to develop a more productive and sustainable agriculture (Riesco et al. 2022), where appropriate culture-dependent bioprospecting campaigns like the present one should be a part of such integrated strategies in order to address critical problems facing humankind, including the need to discovery a new generation of antibiotics to control multidrug-resistant microbial pathogens, bioinoculants to suppress the growth of phytopathogens, and the selection of filamentous actinomycetes that enhance plant growth, alleviate plant stress, reduce reliance on fertilizers and pesticides, and raise soil fertility.
Primary requirements for culture-dependent studies include access to hot spots of actinomycete diversity, associated culture-collections and genomic databases, as well as to chemical libraries of natural products. However, extreme biomes by their very nature are fragile, as witnessed by the destruction of microbial communities in the hyper-arid core of the Atacama Desert following unprecedented rainfall (Azua-Bustos et al. 2018). Consequently, stringent, enforceable measures are needed to protect extreme desert ecosystems, such as those in the Karakum Desert, from fossil fuel and mining interests along the lines promoted by Cockell and Jones (2009) as well as strategies to mitigate climate change. Indeed, it is in our interest as microbiologists to promote microbial conservation as an essential service to humankind.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Glick BR (2012) Plant growth-promoting bacteria: mechanisms and applications. Scientifica 2012. 10.6064/2012/96340110.6064/2012/963401 PMC 382049324278762 · doi ↗ · pubmed ↗
- 2Jiang Y, Li Q, Chen X, Jiang C (2016) Isolation and cultivation methods of Actinobacteria. In Dhanasekaran, D., Jiang, Y., (eds) Actinobacteria–Basics and Biotechnological Applications, London, UK, pp. 39–57
- 3Olson RD, Assaf R, Brettin T, Conrad N, Cucinell C, Davis JJ et al (2023) Introducing the bacterial and viral bioinformatics resource center (BV-BRC): a resource combining PATRIC, IRD and Vi PR. Nucleic Acids Res 51:D 678–D 689. 10.1093/nar/gkac 100310.1093/nar/gkac 1003 PMC 982558236350631 · doi ↗ · pubmed ↗
- 4Saygin H, Nouioui I, Ay H, Guven K, Cetin D, Klenk H-P, Goodfellow M, Sahin N (2020 d) Polyphasic classification of Nonomuraea strains isolated from the Karakum Desert and description of Nonomuraea deserti sp. nov., Nonomuraea diastatica sp. nov., Nonomuraea longispora sp. nov. and Nonomuraea mesophila sp. nov. Int J Syst Evol Microbiol 70:636–647. 10.1099/ijsem.0.00380810.1099/ijsem.0.00380831693475 · doi ↗ · pubmed ↗
- 5Saygin H, Ay H, Guven K, Inan-Bektas K, Cetin D, Sahin N (2021 b) Saccharopolyspora karakumensis sp. nov., Saccharopolyspora elongata sp. nov., Saccharopolyspora aridisoli sp. nov., Saccharopolyspora terrae sp. nov. and their biotechnological potential revealed by genome analysis. Syst Appl Microbiol 44:126270. 10.1016/j.syapm.2021.12627010.1016/j.syapm.2021.12627034653842 · doi ↗ · pubmed ↗