Genomic features and pathogenic potential of Streptococcus agalactiae isolated from bovine clinical mastitis
Jayedul Hassan, Abdus Sattar Bag, Susmita Karmakar, Kishor Sosmith Utsho, Wohab Ali, Ajran Kabir, Tanvir Rahman

TL;DR
This study analyzes the genome of Streptococcus agalactiae from cattle in Bangladesh, revealing its potential to cause disease in both animals and humans.
Contribution
The study identifies novel genomic features and zoonotic potential of S. agalactiae isolates from Bangladesh.
Findings
The isolates belong to rare sequence type ST4 and possess 44 virulence-related genes.
They carry antimicrobial resistance genes and pilus islands linked to invasive diseases.
Source tracking shows the isolates are closely related to human pathogens, indicating zoonotic potential.
Abstract
The goal of this study is to describe the genome of Streptococcus agalactiae that was found in clinical mastitis in cattle in Bangladesh. This work will show how strong the bacteria are and how important they are for public health. Whole genome sequencing (WGS) was performed using the Illumina MiSeq platform, followed by comprehensive analysis with various bioinformatic tools to identify key genomic features. WGS revealed that the isolates are closely related, belonging to sequence type ST4, a rare type previously identified in both human and animal hosts. The isolates possess 44 virulence-related genes linked to adherence, capsule biogenesis, enzyme production, immunoreactive antigens, protease, and cytolysin production. They also carry two pilus islands (PIs), PI-1 and PI-2b, which are often associated with invasive diseases. PI-2b proteins are key targets for vaccine development…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
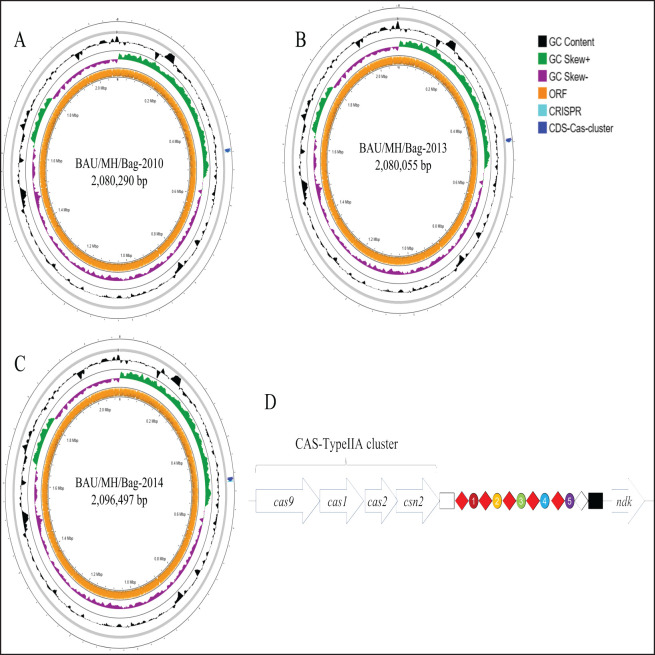 Figure 1
Figure 1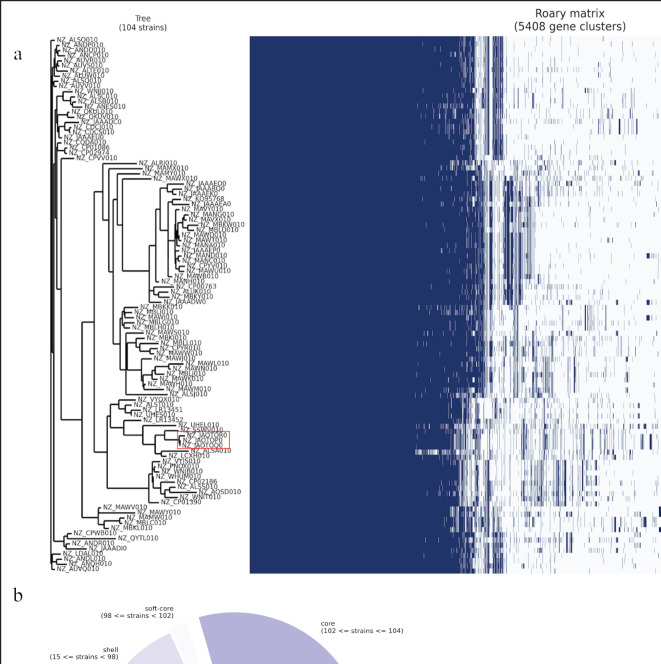 Figure 2
Figure 2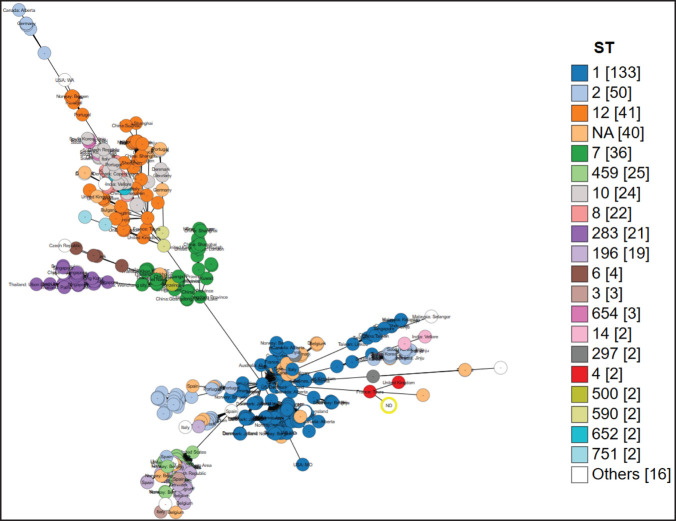 Figure 3
Figure 3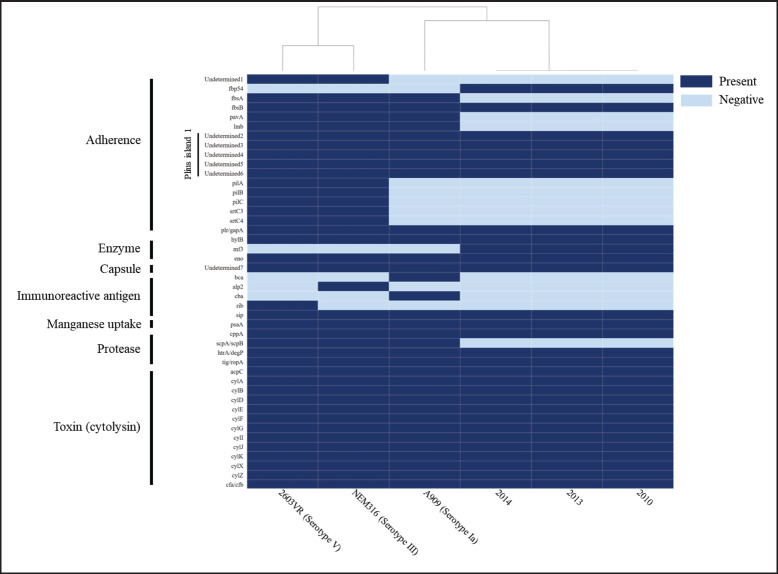 Figure 4
Figure 4 Figure 5
Figure 5 Figure 6
Figure 6| BAU/MH/Bag-2010 | BAU/MH/Bag-2013 | BAU/MH/Bag-2014 | |
|---|---|---|---|
| Bioproject ID | PRJNA879949 | PRJNA879959 | PRJNA879961 |
| Accession No. | |||
| Genome size (bp) | 2080290 | 2080055 | 2096497 |
| GC% | 35.2 | 35.2 | 35.2 |
| No. of contigs | 18 | 20 | 20 |
| Longest contig (bp) | 808064 | 808066 | 807742 |
| Mean contig size (bp) | 115571.7 | 104002.8 | 104824.9 |
| N50 | 534857 | 534857 | 534364 |
| L50 | 2 | 2 | 2 |
| CDS (total) | 2077 | 2074 | 2097 |
| RNAs | 41 [rRNAs: 1, 1, 1 (5S, 16S, 23S); tRNAs: 35; ncRNAs: 3] | 41 [rRNAs: 1, 1, 1 (5S, 16S, 23S); tRNAs: 35; ncRNAs: 3] | 41 [rRNAs: 1, 1, 1 (5S, 16S, 23S); tRNAs: 35; ncRNAs: 3] |
| CRISPR arrays | 1 (Type IIA) - typical | 1 (Type IIA) – typical | 1 (Type IIA) – typical |
| IS elements (no. of sites) | IS3 (4), unknown (5) | IS3 (4), unknown (5) | IS3 (4), unknown (5) |
| ARGs (CARD analysis) | |||
| Sequence type (ST) | ST4 | ST4 | ST4 |
| Virulence genes | 44 | 44 | 44 |
| Strain | Region | Length | Completeness | CDS | Possible phage | GC% |
|---|---|---|---|---|---|---|
| 2010 | 1 | 29.7Kb | Incomplete | 36 | PHAGE_Strept_phiARI0131_2_NC_031941 | 43.20 |
| 2 | 24.9Kb | Incomplete | 8 | PHAGE_Clostr_phiCD27_NC_011398 | 35.79 | |
| 3 | 50.2Kb | Intact | 62 | PHAGE_Strept_Str_PAP_1_NC_028666 | 36.42 | |
| 2013 | 1 | 29.7Kb | Incomplete | 36 | PHAGE_Strept_phiARI0131_2_NC_031941 | 43.20 |
| 2 | 24.9Kb | Incomplete | 8 | PHAGE_Clostr_phiCD27_NC_011398 | 35.79 | |
| 3 | 50.2Kb | Intact | 62 | PHAGE_Strept_315.3_NC_004586 | 36.42 | |
| 2014 | 1 | 29.7 Kb | Incomplete | 36 | PHAGE_Strept_phiARI0131_2_NC_031941 | 43.20 |
| 2 | 24.9 Kb | Incomplete | 8 | PHAGE_Clostr_phiCD27_NC_011398 | 35.79 | |
| 3 | 54.3Kb | Intact | 65 | PHAGE_Strept_315.3_NC_004586 | 36.75 |
| Isolate ID | Pilus Island genes | |||||
|---|---|---|---|---|---|---|
| PI-1 | PI-2a | PI-2b | A | B | C | |
| BAU/MH/Bag-2010 | + | − | + | + | + | − |
| BAU/MH/Bag-2013 | + | − | + | + | + | − |
| BAU/MH/Bag-2014 | + | − | + | + | + | − |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeonatal and Maternal Infections · Milk Quality and Mastitis in Dairy Cows · Oral microbiology and periodontitis research
Introduction
Streptococcus agalactiae, widely known as Group B Streptococcus (GBS), is a gram-positive pathogenic bacterium that causes subclinical and clinical mastitis in dairy cattle, leading to substantial economic losses in the dairy sector [1,2]. Mastitis, an inflammation of the mammary gland, adversely affects milk production and quality, thereby impacting the dairy industry on a global scale. The pathogenic nature of S. agalactiae in bovine mastitis stems from its capacity to adhere to and invade mammary epithelial cells, evade the host’s immune system, and produce various virulence factors (VFs) that intensify the infection [3].
Beyond its veterinary significance, S. agalactiae is a major human pathogen. It is responsible for severe infections such as pneumonia, neonatal sepsis, endocarditis, meningitis, and other serious diseases in humans, particularly affecting newborns, the elderly, and pregnant women [4–6]. The dual-host nature of S. agalactiae raises concerns about its zoonotic potential and the possibility of cross-species transmission. Indirect data suggests that S. agalactiae is transmitted from cattle to humans, posing a significant public health concern [7].
In our previous study, we reported the occurrence of S. agalactiae in clinical mastitis in Bangladesh, highlighting the need for further investigation into its genomic characteristics [8]. In this study, we aimed to provide a comprehensive analysis of the S. agalactiae genomes, focusing on the elucidation of virulence determinants and the public health significance of this pathogen by WGS and analysis.
Considering the findings of previous studies, our research concentrates on several key parameters considered in the molecular epidemiology and genomic diversity of S. agalactiae, such as virulence and antimicrobial resistance (AMR) determinants, sequence types (STs), molecular serotypes, and mobile genetic elements including plasmids, phages, and insertion sequences. Based on our research, this is the inaugural study detailing the genome sequencing and comprehensive analysis of S. agalactiae from cases of clinical mastitis in cattle in Bangladesh. This research not only fills a critical knowledge gap but also provides valuable insights into the pathogenic potential and public health implications of S. agalactiae.
Materials and Methods
Ethical approval
This study does not involve any animals or living beings. Thus, no ethical approval was required.
Bacterial strains
Streptococcus agalactiae was revived from our repository previously isolated from cattle with clinical mastitis [8]. The isolates were recovered from the same dairy farm and were multidrug-resistant with variable resistance patterns.
Sequencing and assembly
Whole genome sequencing (WGS) was conducted on an Illumina NextSeq 2000 platform (Illumina, CA, USA) at the Child Health Research Foundation), Dhaka. Sequence assembly was performed on the Galaxy platform [9]. FASTQ reads were trimmed to filter out low-quality reads on Trimmomatic (Galaxy Version 0.38), followed by assembly by Unicycler (Galaxy Version 0.4.8.0). Following assembly, the genome underwent annotation and ioinformatics analysis to identify STs, virulence genes, and AMR genes (ARGs).
Annotation and bioinformatics analysis
To identify the functional features, annotation was performed by Prokka (Galaxy Version 1.14.6), Rapid Annotation using Subsystem Technology (https://rast.nmpdr.org/rast.cgi), and the NCBI Prokaryotic Genome Annotation Pipeline. Furthermore, the genomes were analyzed with the Roary Pan Genome pipeline for orthologous genes in S. agalactiae and constructed a gene presence-absence matrix [10]. Additionally, the STs of the S. agalactiae genome sequences were determined using the PubMLST website (pubmlst.org). To explore the close relatives, the S. agalactiae sequences were investigated on the BacWGSTdb website (http://bacdb.cn/BacWGSTdb/analysis_single.php) based on core genome multilocus sequence typing (cgMLST). For circular visualization of the S. agalactiae genomes and identification of CRISPR-Cas Proksee tools (https://Proksee.ca) were used.
Mobile genetic elements (plasmids, phages, IS elements)
PlasmidFinder (https://cge.food.dtu.dk/services/PlasmidFinder/) and the PHASTER web server (https://phaster.ca) were used for plasmids and prophages, respectively. For identification of IS elements, the annotated genome was searched on the Isfinder database using default parameters (https://www-is.biotoul.fr/blast.php) as well as CLC Genomic Workbench 22 for manual curation (Qiagen, Germany).
AMR and VF genes
Genes conferring AMR were identified through the Comprehensive Antibiotic Resistance Database (https://card.mcmaster.ca/). For profiling virulence genes, the virulence factor database (www.mgc.ac.cn/VFs) was used.
Pilus island (PI) and gbs2018 genes
Virulence factors related to PI (PI-1, PI-2a, and PI-2b) and highly virulent gene gbs2018 (gbsA, gbsB, and gbsC) were identified in the assembled genome in CLC Genomic Workbench 22 by searching the primer binding sites as described earlier [11–13]. The sequences recovered were further confirmed through a nucleotide homology search (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Moreover, the sequence diversity of gbs2018 genes was determined by alignment and phylogenetic analysis on Mega 11 [14].
Molecular serotyping
Molecular serotyping was performed based on cps gene sequences as described earlier [15]. The nucleotide sequences of CPS*-*encoding clusters were extracted from the S. agalactiae sequences and compared to the previously reported nine distinct types of cps on Easyfig 2.2 software to determine the sequence heterogeneity.
Results and Discussion
Characteristics of the genomes
The S. agalactiae isolates 2010, 2013, and 2014 carried genomes of 2,080,290, 2,080,055, and 2096497 bp, respectively, with a 35.2% GC content (Accession no. JAOTOP000000000, JAOTOQ000000000, JAOTOR000000000) (Table 1). No plasmid-like sequences were identified in the genomes, but they contained incomplete and intact phages as well as insertion sequences of different families (Tables 1 and 2). The general genomic features revealed in this study align with those of S. agalactiae reported earlier [16]. CRISPR/Cas analysis with CRISPR/Cas Finder (https://proksee.ca/) identified a Type-IIA CRISPR array with an associated CAS cluster containing four gene signatures: cas9-cas1-cas2-csn2). The array consisted of 5 identical repeats typical to that reported earlier (5'-GTT TTA GAG CTG TGC TGT TTC GAA TGG TTC CAA AAC-3') [17] and a non-consensus terminal repeat (Fig. 1D). Spacer sequence analysis revealed typical 30 bp spacers, with two spacers (no. 1 and 2) related to prophages and one (no. 3) with dnaC, while the origin of spacers 4 and 5 could not be identified through BLAST search. The Type-IIA CRISPR-cas system is ubiquitous in S. agalactiae and is known as a fully functional CRISPR-Cas system. This system is required for the virulence regulation in S. agalactiae and provides defense against invading genetic elements like phages, plasmids, and transposons [18,19]. Additionally, this system has regulatory effects on the adherence, invasiveness, and biofilm formation by S. agalactiae [20].
MLST and close neighbors
The S. agalactiae isolates belonged to ST4 with unknown clonal complexes through analysis using the PubMLST database. Pan-genome analysis with 104 genomes clustered the isolates 2013, 2014, and 2015 together with minor genetic variations (Fig. 2a), suggesting possible clonality. The occurrence of a clonal strain in the same herd is not impossible. The isolates were closely clustered with human and cow isolates on pan-genome analysis (Fig. 2a), and source tracking through cgMLST analysis revealed close relationships with S. agalactiae reported in humans from various parts of the world (Fig. 3). The genomes consisted of 1,592 core genes within a cluster of 5,408 genes identified in the 104 S. agalactiae genomes (Fig. 2b). Although the genomes clustered with isolates from humans and animals, the closest isolates were from human vaginal samples reported from France and the United Kingdom, which differ by 84 and 156 alleles, respectively. The closest isolates from cow’s milk differed by 613 alleles, suggesting that the study isolates have zoonotic potential. Previous studies have shown that S. agalactiae belongs to different STs with various host specificities [21]. Common strains found in dairy farms around the world are associated with the bovine-adapted ST103, ST568, ST67, ST301, ST313, and ST570 [22], while human isolates belong to ST1, ST7, ST8, ST10, ST12, ST17, ST19, ST23, ST24, ST28, ST110, ST182, ST337, and ST484 [4,23]. The occurrence of ST4 in humans and animals was not readily reported in previous studies and is documented here for the first time in Bangladesh from cases of clinical mastitis in cattle. The close genetic relatedness with both human and animal strains highlights the zoonotic potential of the S. agalactiae strains analyzed in this study.
AMR and virulence factors
Streptococcus agalactiae harbors various virulence factors such as toxins (β-hemolysin/cytolysin and CAMP factor), adhesion and invasion proteins (FbsA, FbsB, αC protein, HylB hyaluronidase, and Rib proteins), resistance elements against antibacterial peptides (βC protein), and mechanisms to evade the host immune systems (production of C5a peptidase and CspA serine protease) [3]. The S. agalactiae genomes analyzed in this study consisted of 44 virulence-related genes, including those associated with adherence (fbp, fbsB, PI-1), capsule biogenesis, enzymes (hylB, eno), immunoreactive antigens (sip), protease (htrA/degP), and toxins (cylABDEFGIJKXZ, cfa/cfb), production, which are considered as the key virulence factors in this bacterium (Fig. 4). These findings indicated that the study strains have the potential to cause mammary gland inflammation and persist in the intra-mammary environment, thereby contributing to mastitis.
In addition to virulence genes, AMR in S. agalactiae poses an additional challenge to controlling mastitis as well as a critical concern to public health [1,2,24]. The study strains possess putative genes encoding resistance to aminoglycosides (orf-BAUMH_00180, BAUMH1_03820, and BAUMH2_00180 of strains 2010, 2013, and 2014, respectively), glycopeptides (vanT, vanY), macrolides (mreA), peptides (mprF), β-lactams, and penicillins (pbp). The genome 2014 also carried the tet(M) gene in addition to that mentioned above (Table 1). The genetic profiles correspond to the observed resistance patterns [8]. However, a difference in the phenotypic resistance was evident between the isolates despite carrying a similar set of resistance genes, highlighting the complexity of AMR. The observed difference might be attributed to the gene expression level, additional resistance mechanisms, gene mutations, and environmental factors. Further genomic and transcriptomic analyses would be necessary to elucidate the underlying cause of these differences. This understanding is crucial to developing effective treatment strategies and mitigating the impact of AMR.
Characteristics of PI and gbs2018 genes
Pilus is an important component of S. agalactiae virulence, facilitating adhesion and attachment to host cells and serving as a potential vaccine target. In S. agalactiae, three PIs have been described, namely PI-1, PI-2a, and PI-2b. S. agalactiae described in this study carried two alleles, PI-1 and PI-2b (Table 3). According to a previous study [13], isolates with PI-1 + PI-2b are frequent in invasive diseases, indicating the study isolates belonged to the invasive subtype. Occurrences of PI-2b have been reported in both human and animal strains with predominance in animal strains [13,23,25,26], and its presence in dairy farms in China and Pakistan suggests its significance in this region [22,27]. Considering the predominance of PI-2b, this study supports the potential of PI-2b protein-based vaccines for S. agalactiae mastitis in both humans and animals, particularly bovines.
The gbs2018 gene encodes a surface adhesin associated with pathogenic GBS strains in humans, animals, and fish. So far, six variants of gbs2018 have been identified with specific host affinities. Variants gbs2018-1 to 3 and 5 were reported from humans and animals, while gbs2018-4 and gbs2018-6 are found in cattle and fish, respectively [28]. Although comprehensive information on the association of different variants with the virulence of S. agalactiae is lacking, variant gbs2018-3 is associated with hyper-virulent ST17 strains reported from humans [29]. The gbs2018-3 was crucial to the hyper-virulence of GBS ST17 clones for adherence and translocation through the intestinal and blood-brain barriers [13]. The study isolates carried gbs2018 A and B genes (Table 3), and the gene sequences were clustered with gbs2018-2 (Fig. 5), a variant reported from a wide range of hosts including human, bovine, canine, feline, and rodents, indicating the potential of the study isolates to adapt in a wide range of hosts and the high potential of transmission between hosts [28].
Circular view of the S. agalactiae isolated from Bangladesh (BD). This figure shows the distribution of ORFs and CRISPR-Cas systems in the circular view (A–C). The linear view of the Cas-cluster in with the distribution of the cas genes, leader sequence (empty rectangle), conserved repeats (red diamond), terminal repeat (white diamond), spacers (different colored ovoids), and the trailer sequence (black rectangle) in S. agalactiae genomes 2010, 2013 and 2014 is shown in panel D. Circular view of the genome and other systems were visualized on Proksee tools (https://proksee.ca/).
Molecular serotype
Molecular serotyping is important for the epidemiological discrimination of GBS, which involves the analysis of cps gene sequences. Previous studies indicated that capsular serotypes may vary among different populations and geographical locations. A total of ten capsular serotypes have been reported in S. agalactiae, with serotypes Ia, Ib, III, and V being more prevalent in the USA and Europe, and serotypes Ia, Ib, II, III, IV, and V are commonly encountered in the South and Southeast regions of Brazil [30]. On the other hand, serotypes VI to IX are sparsely described [31,32]. Thus, determining the capsular serotype is important from an epidemiological point of view as well as for developing a vaccine targeting this potential virulence determinant. Linear comparison on EasyFig revealed that the cps gene clusters in the study isolates were identical to cps-Ia (Fig. 6), aligning with the lineage described in different countries and associated with invasive diseases [33].
Pangenome analysis of the S. agalactiae isolated from Bangladesh (BD). (a) pangenome-based (gene presence and absence) gene clustering matrix of BD (enclosed in red boxes) and isolates from different parts of the world, (b) breakdown of genes in S. agalactiae isolates. The figures were prepared with the data obtained from Roary Pangenome analysis using the roary_plots.py script.
Limitations
The study was limited to three isolates from a single dairy farm in Bangladesh, restricting its ability to provide a comprehensive view of the molecular characteristics of S. agalactiae circulating in the region. Additionally, the genetic characteristics were not validated with phenotypic experiments, limiting the interpretation of genotype-phenotype associations. Thus, further studies with more isolates from different areas of Bangladesh and phenotypic validations are necessary to ascertain the virulence potential and develop effective strategies to control S. agalactiae-associated mastitis and its dissemination to other environmental components, including humans.
Phylogenetic relationship of S. agalactiae isolated in this study and strains reported from different part of the world. The grape tree was prepared on BacWGSTdb website (http://bacdb.cn/BacWGSTdb/) based on core genome multilocus sequence typing (cgMLST) analysis of S. agalactiae with SNP and MLST threshold set to 1,000. Different STs are denoted with range of color schemes and the circle highlighted in yellow indicates isolates described in this study.
Heat map of the virulence factors present or absent in the S. agalactiae genomes from Bangladesh. The virulent genes were identified using VFanalyzer on virulence factor database (VFDB) (www.mgc.ac.cn/VFs). The isolate ID is on the x-axis and the virulence genes/ factors are on the y-axis. Isolates from Bangladesh are marked with an underline. The figure was prepared on DISPLAYR (https://southeastasia.displayr.com/) using default parameters and dendrogram appearance.
Diversity of gbs2018 gene alleles in the S. agalactiae described in this study. Nucleotide sequences of gbs2018 gene alleles were downloaded from GenBank and the evolutionary tree was constructed using the Neighbor-Joining method. The evolutionary distances were computed using the p-distance method and are in the units of the number of base differences per site. This analysis involved 22 nucleotide sequences. All ambiguous positions were removed for each sequence pair (pairwise deletion option). There were a total of 2836 positions in the final dataset. Evolutionary analyses were conducted in MEGA11 [15].
Conclusion
Despite these limitations, this study explored crucial genetic information in S. agalactiae isolated from bovine clinical mastitis in Bangladesh. The isolates belonged to ST4 with a novel clonal complex, carrying virulence genes essential for persistence and pathogenesis in the intra-mammary environment. The isolates belonged to serotype Ia and possess PI-1 and PI-2b, and the gbs2018-2 gene variant, suggesting they are invasive subtypes capable of adapting to diverse hosts, including humans, animals, and rodents. The isolates also harbored multiple AMR genes, highlighting their pathogenic potential and the importance of selecting appropriate antimicrobials for treatment. This study provides a snapshot of S. agalactiae genotypes present in the dairy population of Bangladesh, which is crucial for further studies and suggests vaccine development or importing protein-based vaccines developed against specific virulence determinants such as PI-2b to control S. agalactiae mastitis in cattle in Bangladesh.
Molecular serotyping of S. agalactiae isolated in this study based on capsular polysaccharide (cps) heterogeneity. Nucleotide sequences of cps Ia (LT671983), Ib (LT671984), II (LT671985), III (LT671986), IV (LT671987), V (LT671988), VI (LT671989), VII (LT671990), VIII (LT671991) and IX (LT671992) were downloaded from the GenBank and the linear comparison was performed on Easyfig 2.2. This figure shows the identity of nucleotide sequences of genes associated with capsular polysaccharides.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hernandez L Bottini E Cadona J Cacciato C Monteavaro C Bustamante A Multidrug resistance and molecular characterization of Streptococcus agalactiae isolates from dairy cattle with mastitis Front Cell Infect Microbiol 202111647324 https://doi.org/10.3389/fcimb.2021.6473243399662910.3389/fcimb.2021.647324 PMC 8120232 · doi ↗ · pubmed ↗
- 2Han G Zhang B Luo Z Lu B Luo Z Zhang J Molecular typing and prevalence of antibiotic resistance and virulence genes in Streptococcus agalactiae isolated from Chinese dairy cows with clinical mastitis P Lo S One 2022175 e 0268262 https://doi.org/10.1371/journal.pone.02682623552269010.1371/journal.pone.0268262 PMC 9075616 · doi ↗ · pubmed ↗
- 3Wataradee S Boonserm T Samngamnim S Ajariyakhajorn K Characterization of virulence factors and antimicrobial susceptibility of Streptococcus agalactiae associated with bovine mastitis cases in Thailand Animals 2024143447 https://doi.org/10.3390/ani 140304473833809010.3390/ani 14030447 PMC 10854646 · doi ↗ · pubmed ↗
- 4Furfaro LL Chang BJ Payne MS Perinatal Streptococcus agalactiae epidemiology and surveillance targets Clin Microbiol Rev 2018314 e 0004918 https://doi.org/10.1128/CMR.00049-183011157710.1128/CMR.00049-18PMC 6148196 · doi ↗ · pubmed ↗
- 5Pitts SI Maruthur NM Langley GE Pondo T Shutt KA Hollick R Obesity, diabetes, and the risk of invasive group B streptococcal disease in nonpregnant adults in the United States Open Forum Infect Dis 201856 ofy 030https://doi.org/10.1093/ofid/ofy 0302997795310.1093/ofid/ofy 030PMC 6016410 · doi ↗ · pubmed ↗
- 6Chaguza C Jamrozy D Bijlsma MW Kuijpers TW vande Beek D vander Ende A Population genomics of Group B Streptococcus reveals the genetics of neonatal disease onset and meningeal invasion Nat Commun 20221314215 https://doi.org/10.1038/s 41467-022-31858-43586410710.1038/s 41467-022-31858-4PMC 9304382 · doi ↗ · pubmed ↗
- 7Boonyayatra S Wongsathein D Tharavichitkul P Genetic relatedness among Streptococcus agalactiae isolated from cattle, fish, and humans Foodborne Pathog Dis 202017213743 https://doi.org/10.1089/fpd.2019.26873154986510.1089/fpd.2019.2687 · doi ↗ · pubmed ↗
- 8Hassan J Bag MAS Ali MW Kabir A Hoque MN Hossain MM Diversity of Streptococcus spp. and genomic characteristics of Streptococcus uberis isolated from clinical mastitis of cattle in Bangladesh Front Vet Sci 2023101198393 https://doi.org/10.3389/fvets.2023.11983933753345810.3389/fvets.2023.1198393 PMC 10392839 · doi ↗ · pubmed ↗