Chalcogen Derivatives for the Treatment of African Trypanosomiasis: Biological Evaluation of Thio- and Seleno-Semicarbazones and Their Azole Derivatives
Mercedes Rubio-Hernández, Thaiz R. Teixeira, Tina P. Nguyen, Mai Shingyoji, Elany Barbosa da Silva, Anthony J. O’Donoghue, Conor R. Caffrey, Silvia Pérez-Silanes, Nuria Martínez-Sáez

TL;DR
Researchers designed new compounds to treat African trypanosomiasis by replacing sulfur with selenium, finding some are more effective and less toxic.
Contribution
The study introduces selenosemicarbazone derivatives as more effective and less toxic alternatives to traditional thiosemicarbazones against T. brucei.
Findings
Selenium-based compounds showed better activity against T. brucei than sulfur-based ones.
Selenazole ring structures reduced selenium-related toxicity.
Combining TbrCATL inhibition with antioxidant activity is key for effective treatment.
Abstract
Human African trypanosomiasis (HAT) is caused by Trypanosoma brucei. Drug therapy remains challenging due to drug resistance and/or toxicity. New drugs are needed. Using thiosemicarbazones as a starting point, we employed a S to Se isosteric replacement strategy to design 44 analogs which were evaluated against T. brucei in vitro. Compounds were divided into 11 groups of four derivatives corresponding to thio-, selenosemicarbazones, and their cyclic counterparts, thio- and selenazoles. We selected three groups which contained a total of six derivatives that inhibited parasite growth by >70%. Then, we investigated the mechanism of action of these compounds, performing quantitative assays to measure their inhibition of the T. brucei cathepsin L-like protease (TbrCATL) and DPPH antioxidant activities. The lead compound (SeO3) showed antioxidant capacity and the best activity against T.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1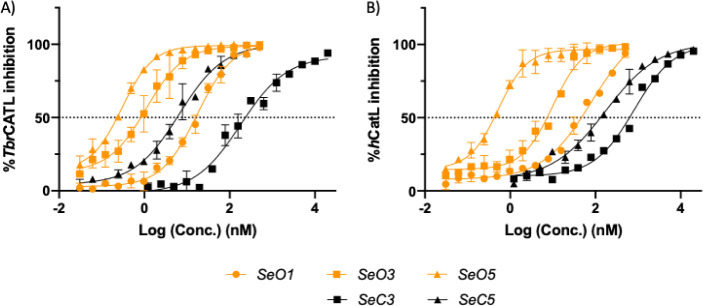 2
2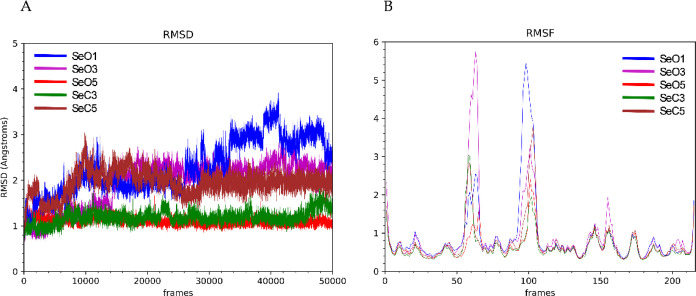 3
3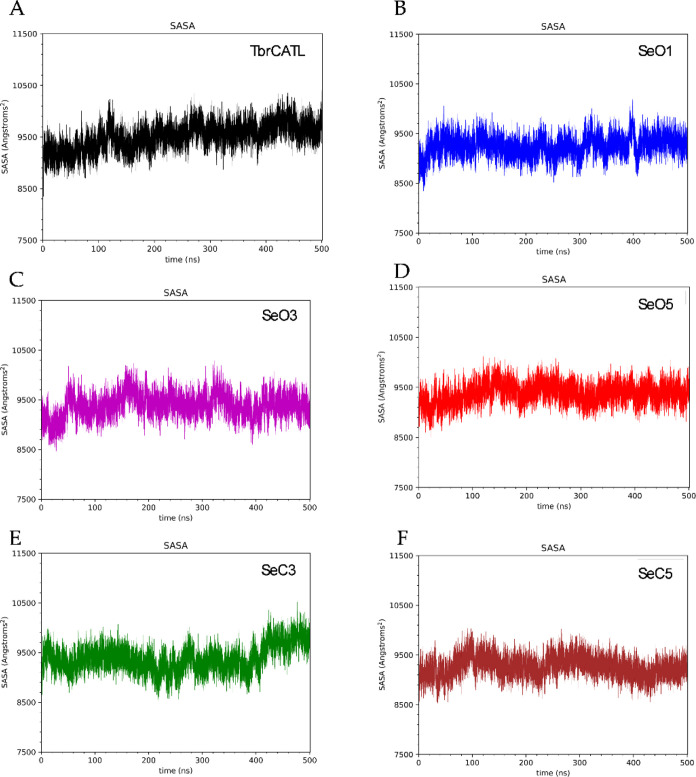 4
4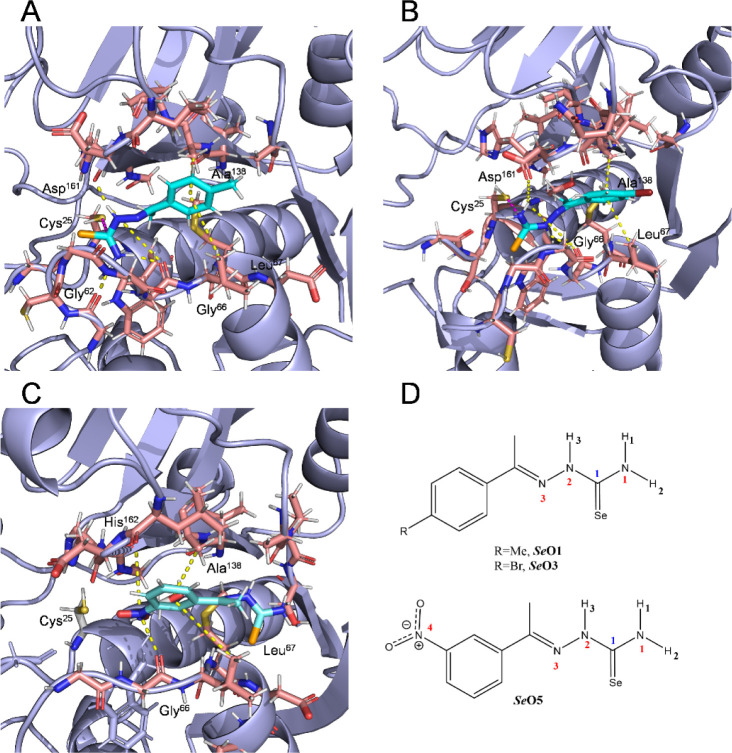 5
5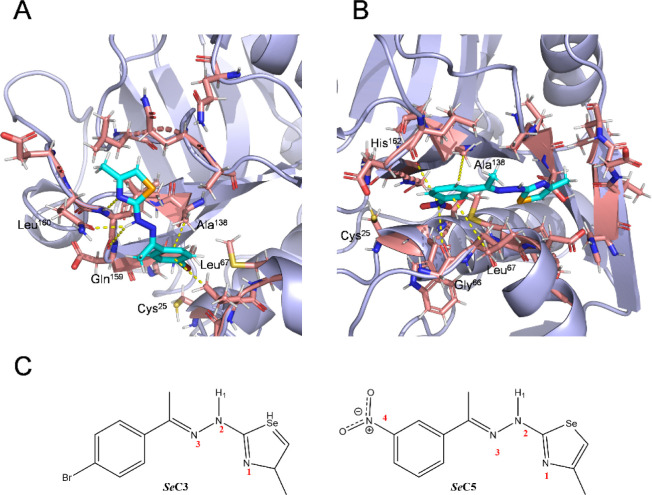 6
6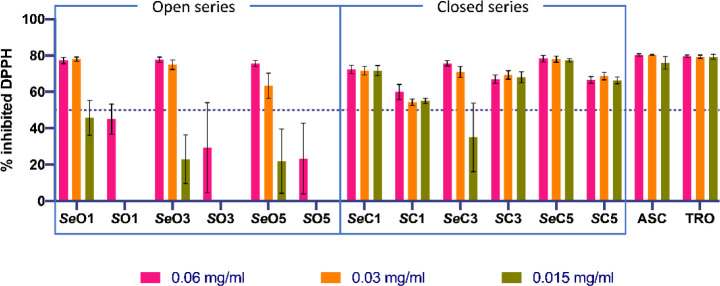 7
7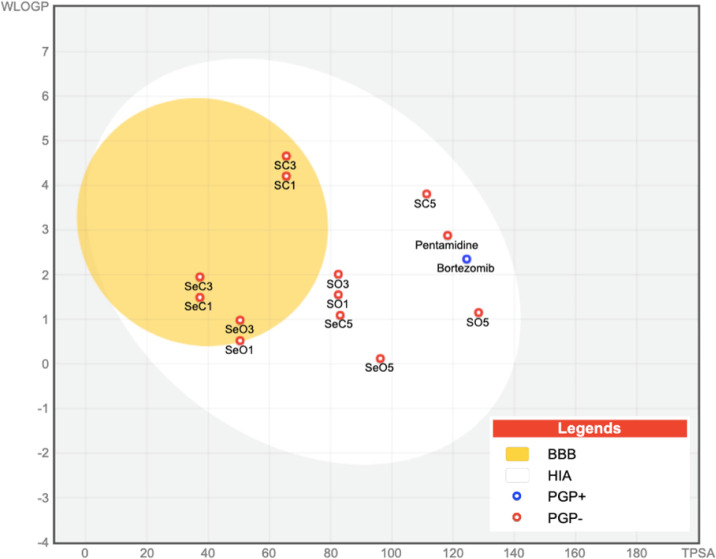 8
8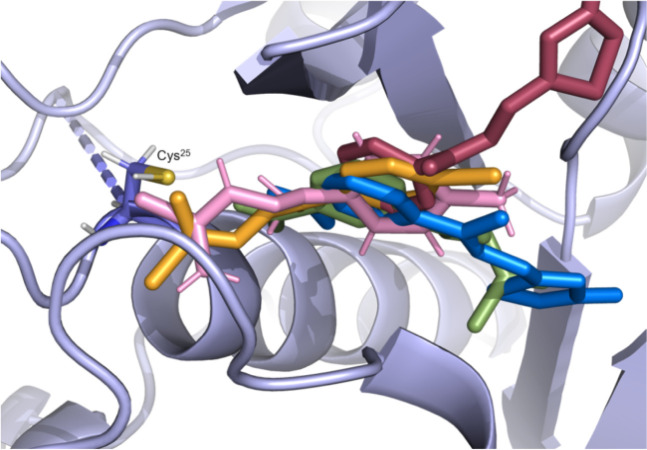 9
9| Compound | IC50
| IC50
| SI
| |
|---|---|---|---|---|
| 97.08 | 17.90 ± 3.62 | 84.52 ± 17.03 | 5 | |
| 51.07 | ND | ND | ND | |
| 79.98 | ND | ND | ND | |
| 32.85 | ND | ND | ND | |
| 99.15 | 1.22 ± 0.52 | 9.87 ± 0.63 | 8 | |
| 101.02 | 201.65 ± 0.92 | 792.75 ± 66.54 | 4 | |
| 95.54 | 7.33 ± 0.27 | 135.95 ± 19.87 | 19 | |
| 53.99 | ND | ND | ND | |
| 101.47 | 0.30 ± 0.07 | 0.51 ± 0.08 | 2 | |
| 98.02 | 6.91 ± 2.64 | 174.70 ± 43.70 | 25 | |
| 100.00 | 5.83 ± 0.35 | 20.74 ± 4.36 | 4 | |
| 87.70 | 82.57 ± 4.05 | 4065.50 ± 382.54 | 49 | |
| E-64 | ND | 3.1 ± 0.0e | 30.0 ± 4f | 10 |
| System | ΔG (kcal/mol) | SASA (Å) |
|---|---|---|
| N/A | 9504.6 ± 248.1 | |
| –9.32 ± 0.33 | 9251.1 ± 200.7 | |
| –6.72 ± 0.36 | 9380.7 ±230.1 | |
| –5.37 ± 0.51 | 9374.2 ± 195.5 | |
| –4.85 ± 0.30 | 9378.3 ± 255.4 | |
| –5.37 ± 0.51 | 9261.4 ±198.9 |
- —National Institute of Allergy and Infectious Diseases10.13039/100000060
- —University of California, San Diego10.13039/100007911
- —Universidad de Navarra10.13039/501100004435
- —Gobierno de Navarra10.13039/501100017266
- —Ministerio de Educaci?n y Formaci?n Profesional10.13039/501100020636
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTrypanosoma species research and implications · Research on Leishmaniasis Studies · Synthesis and Biological Evaluation
Introduction
Human African trypanosomiasis (HAT; aka sleeping sickness) is a neglected tropical disease (NTD) that is caused by Trypanosoma brucei, a protozoan parasite that infects humans in Sub-Saharan Africa. HAT is caused by two subspecies of T. brucei, Trypanosoma brucei gambiense and Trypanosoma brucei rhodesiense, which are transmitted by the bite of the tsetse fly (Glossina spp.). Most cases (92%) are due to T. b. gambiense, which is prevalent in west and central of Africa, and develops as a chronic, and ultimately, fatal disease that affects the central nervous system (CNS). T. b. rhodesiense (8% of cases) is endemic in southern and eastern parts of Africa, and causes a more acute, but likewise, fatal, CNS disease. ?,? In livestock, particularly cattle, Trypanosoma brucei brucei is one of a number of African trypanosomes responsible for Nagana disease,? while also being used as a laboratory model for both of the human-infective T. brucei subspecies.
To treat HAT, the few medicines available suffer from toxicity and administration routes that complicate treatment (intramuscular for pentamidine or intravenous for melarsoprol, suramin and eflornithine) in resource-poor medical environments.? Moreover, the choice of drug depends on the stage of the disease diagnosed and the causative agent. Specifically, pentamidine and suramin are used for first (hemolymphatic) stage infections of T. b. gambiense and T. b. rhodesiense, respectively; whereas melarsoprol, and nifurtimox combined with eflornithine, are utilized to treat the second (CNS-infiltrated) stage of disease due to T. b. gambiense and T. b. rhodesiense, respectively.? Resistance, including cross-resistance, has been documented for melarsoprol, ?−? ? pentamidine? and eflornithine.? Since its approval in 2018, the oral drug, fexinidazole, has been recommended for both stages of the disease regardless of infecting subspecies, however, this drug has side effects. ?,? Overall, there is a need to discover new orally administered alternatives with acceptable safety profiles.
Recent advances in the search for pharmacological therapies have included the design of inhibitors of a cysteine protease known as T. brucei cathepsin L (TbrCATL; formerly known as rhodesain. ?,? ) TbrCATL is the major papain family, cysteine protease found in T. brucei.? This enzyme contributes to the parasite’s ability to cross the blood-brain barrier (BBB) that facilitates the second stage of HAT.? The protease also participates in various processes,? such as nutrition,? host cell invasion? and differentiation.? TbrCATL is homologous to cruzain (Cz) in Trypanosoma cruzi, the etiological agent of Chagas disease.? Cz and TbrCATL are validated drug targets ?,? and share 70% of the sequence, and differing by just two amino acids in the active site. ?,? Therefore, compounds that inhibit Cz and T. cruzi growth, ?,? might also demonstrate activity against TbrCATL and T. brucei. Previously, synthetic compounds inhibiting TbrCATL by >99% have shown trypanocidal activity. ?,?
Thiosemicarbazones (S-semicarbazones) and thiazoles have demonstrated activity against T. brucei and inhibit TbrCATL ?,?−? ? ?
S-semicarbazones are particularly suitable for the treatment of NTDs because of their low-cost synthesis, low molecular weight and nonpeptidic nature. They are also covalent reversible inhibitors of cysteine proteases, and serve as intermediates in the synthesis of thiazoles ?,?−? ? ? Given these benefits, we investigated the isosteric replacement strategy of changing sulfur (S) to selenium (Se),? to synthesize the Se-counterparts of S-semicarbazones and thiazoles, which are, respectively, selenosemicarbazones (Se-semicarbazones) and selenazoles. S and Se both belong to the chalcogen group, and they are bioisosteres, thus, their physicochemical properties are very similar. However, because Se is slightly bigger, the valence electrons are loosely bonded, increasing the reactivity of Se and making it more nucleophilic and polarizable compared to S. The increased reactivity of Se contributes to its electronegativity and redox properties, ?,? which are imparted to the molecules that contain this element. ?−? ?
Se is a trace element essential for both parasite and human survival, however, overdosing can lead to toxicity. This element stimulates the immune system and is a key component in selenoproteins, such as glutathione peroxidase and selenocysteine.? Se deficiency does not directly cause disease but weakens the organism to facilitate various pathological conditions. ?,?,? Compounds containing Se have been explored to treat different parasitoses like Chagas disease ?,? and leishmaniasis.? However, data for Se-containing small molecules against T. brucei are scarce. To date, only the anti-inflammatory and antioxidant compound, ebselen, is known to inhibit T. brucei hexokinase 1 ?,? and trypanothione synthetase,? two enzymes that participate in glycolysis? and in the defense against the oxidative damage,? respectively. In addition, an in vivo study showed that T. brucei-infected rats improved their response to the disease when supplemented with dietary Se.? Consequently, we consider the isosteric replacement of S with Se an interesting strategy in the pursuit of new therapies to treat HAT.
Bearing in mind previous reports which demonstrate anti-T. brucei activity and inhibition of TbrCATL, ?,?−? ? ? as well as our previous research which showed that selenazoles are active against T. cruzi and inhibit Cz? (Figure A,B), we evaluated two series of compounds (* S * O, * Se * O, * S * C, * Se * C) against T. brucei (FigureC). The first series contained “open” S-semicarbazone (* S * O) and Se-semicarbazone (* Se * O) derivatives. The second series comprised the corresponding cyclic or “closed”, thiazole (* S * C) and selenazole (* Se * C) counterparts (Figure). With these series, our objective here was to identify a compound(s) that selectively kills T. brucei and inhibits TbrCATL. Of note, this is the first time that synthetic organic seleno-derivatives have been tested against T. brucei.
*Design strategy. (A) Compounds previously shown to have activity against T. brucei and TbrCATL ,, and (B) T. cruzi and cruzain. (C) General structures of the compounds studied: S-semicarbazones ( S
O), Se-semicarbazones ( Se
O), thiazoles ( S
C) and selenazoles ( Se
C).*
Results
Biological Evaluation
The activity of 44 compounds (* S * O1 -S O11, * Se * O1- Se O11, * S * C1- S C11, * Se * C1- Se C11) was evaluated against T. brucei brucei Lister 427 in vitro.? First pass screens at 10 μM identified 12 actives (Table S1) that inhibited parasite growth by >70%. These 12 compounds were selected for dose response (DR) analysis, as well as for DR counter cytotoxicity assays using human embryonic kidney (HEK)293 and hepatoblastoma (Hep)G2 cells (Table S1), both of which are commonly used to assess compound toxicity. All 12 compounds showed >50% cytotoxicity for both cell lines (Table S1), but the concentrations at which cell growth was inhibited by 50% (CC_50_ values) were at least twice as high for the selenazoles as for the Se-semicarbazones (Table S1). * Se * O3 was the most potent compound with an EC_50_ value (concentration at which parasite growth is inhibited by 50%) of 0.47 μM. Relative to the CC_50_ values of 2.70 and 2.82 μM obtained with the HEK293 and HepG2 cells, respectively, the selectivity index (SI) for * Se * O3 was the highest of the compounds tested with a value of approximately 6.
Based on the parasiticidal and toxicity data, we selected the Se-compounds, * Se * O1, * Se * C1, * Se * O3, * Se * C3, * Se * O5, * Se * C5 for additional assays (see below). We also included the corresponding S-counterparts (* S * O1, * S * C1, * S * O3, * S * C3, * S * O5, * S * C5) for comparison. Table shows the biological data for this top 12 hits.
1: Biological Data for the Top 12 Hits against T. brucei, and HEK293 and HepG2 Cells
Mechanism of Action: Enzyme Inhibition Assays
We conducted enzyme inhibition assays with TbrCATL and the selected top 12 compounds (Table) to gain deeper insight into their possible mechanism of action (see Table). These assays also enable a direct comparison of structural and functional differences between the S and Se compounds as well as the open and closed compounds. First pass screens at 10 μM identified eight compounds that inhibited the activity of TbrCATL by >85% (Table). Of these eight, five were Se-compounds ( Se * O1, * Se * O3, * Se * C3, * Se * O5 and * Se * C5) and three were S-compounds ( S * O3, S O5 and * S * C5). These compounds were subjected to DR analysis to measure the IC_50_ values (i.e., the concentration of compound necessary to inhibit 50% of the enzyme’s activity). In parallel, and to assess compound selectivity for TbrCATL, we measured their IC_50_ values for inhibition of the orthologous human cathepsin L (hCatL; Table). The DR data for the five *Se-compounds against both enzymes are shown in Figure. We note the shift to the left of the IC_50_ values for the open compounds relative to their cyclic derivatives, especially for * Se * O3 which was 100 and 300 times better than * Se * C3 against TbrCATL and hCatL, respectively. The DR data for the S-compounds shows the same shift to the left (Figure S1). Nevertheless, the IC_50_ values of the compounds within the same series ( Se * O1, * Se * O3, * S * O3, * Se * O5, * S * O5) are similar and independent of the chalcogen atom present in the molecule (Table). Based on the selectivity of inhibition (i.e., IC_50_ hCatL/IC_50_ TbrCATL), the best compound was * S * C5 (SI = 49), a *S-*derivative that does not have activity against T. brucei. Of the compounds shown in Tables and ?, * Se * O3 stood out for combining activity against T. brucei with inhibition of TbrCATL (EC_50_ = 0.47 μM and SI_ TbrCATL_ = 8).
2: Inhibition of TbrCATL and hCatL by the Top 12 Anti-Trypanosomal Agents
DR data for Se compounds against TbrCATL (A) and hCatL (B).
Mechanism of Action: Molecular Dynamics (TbrCATL)
To study the interactions of * Se * O1, * Se * O3, * Se * O5, * Se * C3, and * Se * C5 with TbrCATL, molecular dynamics (MD) simulations were conducted. Our aim in this assay is to better understand the differences in the interactions between the open and cyclic *Se-*compounds. The ligand, K11777 (a covalent irreversible cysteine protease inhibitor), in the 2P7U? crystal structure was replaced with the different Se derivatives prior to running the MD simulations. A 500 ns trajectory was produced and analyzed to evaluate the stability of the complexes. Root mean square deviation (RMSD) values and root-mean-square fluctuation (RMSF) profiles were obtained (Figure). RMSD analysis indicated that the protein reaches structural stabilization in the complex at different times during the simulations depending on the ligand. The TbrCATL in complex with * Se * O1 remained stable from 120 to 260 ns, with a mean RMSD value of 2.5 Å. In the brominated derivative * Se * O3 complex, the protein required 150 ns to stabilize, exhibiting a RMSD value of 1.93 Å, whereas its closed analog, * Se * C3, the enzyme presented lower deviations from 60 ns to the end of the trajectory (1.19 Å). The protease in complex with * Se * O5 was rapidly stabilized within 20 ns achieving a mean RMSD value of 1.09 Å, whereas with the closed nitro derivative, * Se * C5, stabilization occurred at 150 ns with a mean RMSD value of 1.94 Å. The RMSF profiles demonstrated a similar pattern of residue fluctuations across all five complexes.
*(A) Plot of the evolution of RMSD values during MD simulations of TbrCATL complexed with the ligands Se
O1 (blue), Se
O3 (purple), Se
O5 (red), Se
C3 (green) and Se
C5 (brown). (B) RMSF plot of protein residues in complex with the ligands Se
O1 (blue), Se
O3 (purple), Se
O5 (red), Se
C3 (green) and Se
C5 (brown).*
The solvent-accessible surface area (SASA) study suggested that the binding of the five ligands reduced the solvent-accessible surface area of the unbound TbrCATL (Table). Also, the SASA graphs exhibited minimal fluctuations throughout the simulation trajectories (Figure). These findings align with the molecular mechanics Poisson–Boltzmann surface area (MMPBSA) binding free energy calculations, which yielded consistently negative values for all complexes (Table), thus, confirming thermodynamically favorable interactions between TbrCATL and the Se-based ligands.
*Solvent accessible surface area (SASA) of unbound TbrCATL (A) and TbrCATL complexed with Se
O1 (B), Se
O3 (C), Se
O5 (D), Se
C3 (E) and Se
C5 (F).*
3: Predicted Binding Free Energy Values and Mean SASA Values Obtained by MD Simulations
The interaction map of the open ligands * Se * O1, * Se * O3, and * Se * O5 with TbrCATL is shown in Figure. Compound * Se * O1 forms hydrogen bonds through the selenosemicarbazone group hydrogens. Specifically, H3 interacts with the carbonyl oxygen of Asp^161^ and Gly^66^, while H1 forms polar contacts with Gly^66^ and H3 with Gly^62^ (see FigureD for ligand atom identification). Additionally, CH-π interactions are observed between the side chains of Leu^67^ and Ala^138^ at the S2 pocket and the aromatic ring of the ligand. A similar interaction pattern is displayed by * Se * O3, which also forms polar contacts through its H1 and H3 with Asp^161^ and Gly^66^, as well as CH-π interactions via its bromophenyl group with Leu^67^ and Ala^138^. In contrast, * Se * O5 does not form any hydrogen bond with the protein (Figure). However, it still exhibits CH-π interactions through its aromatic ring and additional π-hole interactions between the electron-deficient nitrogen of the nitro group and the carbonyl groups of Gly^66^ and His^162^ (Table S2 for average distances of these interactions along with their occurrence during the simulation time).
*Representative frames of the binding modes for Se
O1 (A), Se
O3 (B) and Se
O5 (C) in the TbrCATL active site (PDB: 2P7U) obtained from MD simulations. The frames selected here show the predominant conformations of the ligand-protein complex throughout the simulation. Yellow dashed lines indicate all interactions between the ligands and the protein, including hydrogen bonds, CH-π interactions and π-hole interactions. Pink dashed lines highlight the potential covalent bond formed between the thiol group of Cys25 and the selenocarbonyl group of the ligands. (D) Ligand atom labels involved in the recognition process.*
*Representative frames of the binding modes of compounds Se
C3 (A) and Se
C5 (B) at the TbrCATL active site (PDB: 2P7U), obtained from MD simulations. The frames selected here show the predominant conformations of the ligand-protein complex throughout the simulation. Yellow dashed lines indicate all interactions between the ligands and the protein, including hydrogen bonds, CH-π interactions and π-hole interactions. (C) Ligand atoms labels involved in the recognition process.*
Mechanism of Action: Radical Scavenging Capacity–Antioxidant
Activity
As a possible alternative or additional mechanism of action, we evaluated the antioxidant capacity of the compounds shown in Tables and ? at three different concentrations (0.06, 0.03, and 0.015 mg/mL), using the DPPH assay.? The assay was designed to measure the ability of the compounds to act as free radical scavengers or hydrogen donors. Quantitative data are shown in Table S3, and the graphical representation of the maximal antioxidant activity after 2 h is shown in Figure.
Antioxidant activity of the compounds of interest at the three tested concentrations after 2 h. ASC, ascorbic acid and TRO, trolox, were used as positive controls.
All compounds, except * S * O1, * S * O3 and * S * O5, scavenged free radicals from 1,1-diphenyl-2-picrylhydrazyl (DPPH) by >50% after 2 h at the highest concentration tested (0.06 mg/mL). Therefore, these three compounds were not tested further (Table S3). Compared to the chalcogen counterparts, the Se compounds demonstrated enhanced antioxidant activity (70–80%) at 0.06 and 0.03 mg/mL relative to their S analogues (40–70%), especially the open series, for which the S compounds failed to reach the 50% free radical inhibition threshold. This improved antioxidant activity by the Se compounds may be attributed to the higher reactivity of Se. In contrast, for the cyclic derivatives, the structure itself may enhance antioxidant activity, suggesting that the cyclic configuration plays a role alongside the chalcogen element. In this context, * Se * C1 and * Se * C5, maintained similar inhibition values at the three concentrations tested (Figure). Indeed, * Se * C5 is outstanding for its remarkable activity, which was comparable to that of the positive controls, ascorbic acid (ASC) and trolox (TRO).
In Silico prediction of
ADME and Drug-Likeness Properties
To predict their ADME and drug-likeness properties, all 44 compounds, and the positive drug controls, pentamidine and bortezomib, were analyzed using the SwissADME platform (http://www.swissadme.ch/).[?](#ref50) Data regarding molecular weight, lipophilicity, solubility, drug elimination and drug likeness criteria are available in Table S4.
None of the 12 top hits (Tables and ?) violated any of the drug-likeness criteria (Lipinski, Ghose, Veber and Egan rules) available on SwissADME platform (Table S4). In addition, all hits showed adequate oral bioavailability, scarce inhibition of cytochrome P450 (Table S4) and good gastrointestinal absorption (HIA; Figure). Also, we would note that that the cyclic derivatives, * Se * C1, * Se * C3, * S * C1 and * S * C3, are more likely to cross the BBB than their open analogues (Figure). Data regarding passive gastrointestinal absorption (HIA) and brain access (BBB) are shown in the BOILED-Egg graph? (Figure). From this graph, we can also ascertain that only bortezomib (shaded in blue) is a substrate for P-glycoprotein (PGP+), a key protein responsible for efflux through biological membranes, giving us an idea of the mechanism of permeability used by our compounds.
BOILED-Egg model of the 12 top hits and positive drug controls (pentamidine and bortezomib). The white region contains those compounds that are prone to be passively absorbed from the gastrointestinal tract (HIA). The yellow region (yolk) contains the molecules predicted to cross the BBB. Compounds colored in blue are predicted to be P-glycoprotein substrates (PGP+), whereas those in red are not (PGP−).
Discussion
Se has been neglected in medicinal chemistry, mainly because of its toxicity? and the comparison of S- and *Se-*containing compounds has been scarce. Accordingly, we implemented an isosteric replacement strategy for semicarbazones by changing S to Se. S-semicarbazones are the starting point for this work because they are established inhibitors of the trypanosomal cysteine proteases, Cz and TbrCATL [28–30]. We also synthesized thiazoles and selenazoles to include structural variability and, for the first time, test selenazoles against T. brucei. Of 44 novel chalcogen derivatives tested against T. brucei, the Se derivatives were the most active (>70%). Structurally, nine of the active compounds ( Se * O1, * Se * O2, Se O3, * Se * O4, * Se * O5, * Se * O6, * Se * O9, * Se * O10, * Se * O11) belong to the open series, whereas the remaining three ( Se * C1, * Se * C3, * Se * C5) are closed derivatives. Comparing the EC_50_ values of these Se-derivatives, the open compounds ( Se * O3 and * Se * O1) are at least ten times better than their corresponding cyclic analogues ( Se * C3 and * Se * C1). Overall, the combination of the open conformation and Se improves activity against T. brucei, as demonstrated by the best compound, * Se * O3 and its EC_50_ value of 0.47 ± 0.02 μM.
Regarding toxicity, 19 of the 22 *Se-*compounds were toxic, in contrast to five S-compounds. All of the trypanocidal Se-derivatives were cytotoxic to both HEK293 and HepG2 cells with similar CC_50_ values and SIs (Table S1). We only performed DR analysis for those compounds that were active against T. brucei. Of these, * Se * O3 had the best SI_HEK293_ value of 6. Nevertheless, we found that the cyclic *Se-*derivatives were less toxic than their open counterparts. This is in accordance with other studies in the literature which report that thiazoles decrease toxicity ?−? ? ? This suggests that the structural configuration of the cyclic derivatives plays a key role in mitigating the inherent toxicity of Se, potentially due to increased stability. This finding is also consistent with our previous data published for C2C12 cells (immortalized mouse myoblasts),? and it will be necessary to design less toxic derivatives.
Regarding the possible mechanism of action, as we reported for the main cysteine protease of T. cruzi, Cz,? the open compounds are the more potent inhibitors of TbrCATL (Figure). This is independent of the chalcogen compound present in the molecule, as the IC_50_ values between S- and Se-derivatives are very similar (0.30 to 17.90 nM). In addition, the nitro group in the aromatic ring may influence the interaction with TbrCATL because all derivatives that contain this group (* Se * O5, * Se * C5, * S * O5 and * S * C5) inhibit the protease. For these reasons, we decided to investigate the interactions between the ligands and TbrCATL utilizing MD simulations.
We performed MD simulations to determine the binding mode of * Se * O1, * Se * O3, * Se * O5, * Se * C3 and * Se * C5 with TbrCATL. This involved studying the interactions between the protease and the ligands, and analyzing the potential formation of a covalent bond between the selenosemicarbazone moiety of open derivatives and the catalytic Cys^25^, as previously described. ?,? The interaction maps for * Se * O1 (FigureA) and * Se * O3 (FigureB) indicates a common mode of recognition and binding to the TbrCATL active site. The aromatic ring of both compounds participates in CH-π interactions and adopts a conformation whereby the selenosemicarbazone moiety forms multiple hydrogen bonds with various residues in the binding site. This ligand disposition satisfies the optimal geometric parameters necessary to potentially enable a nucleophilic attack by Cys^25^ on the selenocarbonyl group, leading to the formation of a covalent bond. This recognition mode is similar to that exhibited by structurally related S-semicarbazones previously described.? Of note, although the bromine-substituted closed derivative (* Se * C3) displays the same pattern of CH-π interactions via its aromatic ring (FigureA), the selenazole counterpart points outward from the binding site, forming a different network of hydrogen bonds. Both polar and nonpolar interactions are present in lower occurrences during the trajectory of the molecular dynamics (Table S2). Differences in contacts and the potential for covalent bond formation could explain the greater enzyme inhibition observed for the open compounds compared to their closed analogues (* Se * O3 vs SeC3). In addition to this information, the RMSD study of the * Se * O1 and * Se * O3 complexes shows greater deviation during the trajectory, likely due to the orientation of the selenosemicarbazone moiety toward the catalytic Cys^25^. This orientation may result in a decreased stabilization of the protein in both complexes, although it potentially facilitates the formation of a covalent bond.
The analysis of the interactions of * Se * O5 (FigureC) and * Se * C5 (FigureB) with TbrCATL indicates that the nitro group of the nitrophenyl substituent plays a crucial role in enzyme inhibition. The electron-deficient nitrogen of the nitro aromatic group participates in π-hole interactions.? These polar interactions place the aromatic ring of the ligands in the same region of the binding site as the phenyl group of * Se * O1 and * Se * O3, establishing CH-π interactions (Figure). The interactions involving the nitro group forces similar conformations for the open and closed chains of both ligands. The selenosemicarbazone chain of * Se * O5 adopts a conformation that positions the selenocarbonyl group away from Cys^25^, preventing both covalent bond formation and polar interactions between the selenosemicarbazone hydrogens and the enzyme. The orientation of the chain containing the selenazole of * Se * C5 matches the arrangement found in * Se * O5, underscoring the role of the nitro group in the binding mode of both ligands. This configuration of the nitro-aromatic derivatives aligns with our previous findings,? in which interactions between * Se * C5 and Cz were analyzed. Predictions for * Se * C5 show that it interacts with Cz through a similar π-hole pattern as found for TbrCATL, which is expected due to the structural similarities between both cysteine proteases. ?,?,? These data align with the enzyme inhibition data, which show similar IC_50_ values for the open and closed derivatives bearing the nitro substituent (Table). Collectively, these findings provide valuable structural insights for the future design of compounds that effectively inhibit both TbrCATL and Cz.
*Superposition of the structures of Se
O1 (pink), Se
O3 (orange), Se
O5 (green), Se
C3 (red) and Se
C5 (blue) in the TbrCATL active site (PDB 2P7U) obtained from MD simulations. The frames selected here show the predominant conformation of each ligand bound to the protein throughout the simulation.*
In addition to the possible mechanism of TbrCATL inhibition, the data generated for the trypanocidal activity suggest the presence of an additional mechanism(s) of action. This hypothesis is supported by the observation that the most potent trypanocide, * Se * O3, inhibits TbrCATL (1.22 nM), whereas * Se * C1, which is active against T. brucei, does not achieve >85% inhibition of the protease. This might be due to differences between the cell environment and the inhibition assay conditions. For T. brucei, redox homeostasis is critical to survival within the oxidative environment of the host.? Unlike mammalian cells, it lacks catalase and classical glutathione-based detoxification pathways, instead depending primarily on the trypanothione system to manage oxidative stress.? This unique redox system represents a key vulnerability that can be therapeutically exploited. ?−? ? Studying the antioxidant activity of anti-T. brucei compounds provides valuable insights into their ability to interfere with the parasite’s redox balance. Compounds that induce oxidative stress or disrupt redox homeostasis may selectively impair parasite viability without significantly affecting host cells, while the antioxidant properties could also modulate parasite survival under stress conditions. Therefore, evaluating the redox behavior of candidate molecules not only contributes to an understanding of their mechanism of action but also aids in identifying redox-active compounds with selective antiparasitic potential. ?,? In this context, selenium is well-known for its antioxidant properties, ?,? which led us to hypothesize that selenium-containing compounds may retain this activity and potentially influence T. brucei metabolism.
The parasite generates reactive oxygen species (ROS) and utilizes them as part of its defense system,? suggesting that modulation of oxidative processes could alter its viability. Accordingly, with the compounds from the groups shown in Tables and ?, we performed the DPPH assay to measure antioxidant activity, in the knowledge that Se is known for its free radical-scavenging activity. ?,? The data reveal that the main contributor to the antioxidant activity measured is the structure. Specifically, all cyclic compounds were antioxidants, whereas only the Se-compounds from the open series acted as such (Figure). This suggests a direct correlation between the structural configuration and the stability of Se within the compounds such that the greater stability of the cyclic structures facilitates sustained antioxidant activity.
We suggest that both possible mechanisms of action are important to consider when designing future trypanocidal Se-containing compounds. This notion is reinforced by the fact that the derivatives, * Se * O1, * Se * O3, * Se * O5, * Se * C3 and * Se * C5 inhibit TbrCATL between 0.30 and 17.90 nM (Table) and possess >70% antioxidant activity at 0.06 and 0.03 mg/mL (Figure). If one of these conditions is missing, the compound does not affect the parasite, e.g., * Se * C1, which shows antioxidant activity but does not inhibit TbrCATL.
Se-semicarbazone- and selenazole-based derivatives are also bioactive against T. cruzi, and inhibit that parasite’s major cysteine protease, Cz.? Indeed, a trend for inhibition of both TbrCATL and Cz can be observed (see Table S5), with the Se-containing derivatives standing out as being the most active. The similarity in inhibition profiles suggests that the Cz-based design strategy employed in that previous work has been successfully extended to TbrCATL, supporting the development of broader-spectrum antiparasitic agents that target both Chagas disease and HAT.
A comparative analysis of the SAR revealed that cyclic selenazole-based derivatives are generally less toxic against T. cruzi and T. brucei, than their open-chain counterparts and exhibit greater antioxidant potential (See Table S5). However, in terms of enzyme inhibition, the Se-semicarbazone derivatives generate lower IC_50_ values for both Cz and TbrCATL, aligning with their reported covalent inhibition mechanism of Cz.? The molecular dynamics simulations performed here also support covalent inhibition, indicating that Se-semicarbazones adopt conformations that favor a nucleophilic attack by the catalytic Cys^25^ in TbrCATL.
Further structural studies revealed that although cyclic derivatives display higher IC_50_ values (See Table S5), likely due to their inability to form a covalent bond, they still engage in strong interactions with the proteases via the aromatic ring, especially those compounds bearing substituents like the NO_2_ group (* Se * O5 and Se C5) (See FiguresC and ?B). Interestingly, the binding mode of * Se * C5 in the active site of TbrCATL mirrors that observed with Cz,? suggesting a consistent interaction pattern across both enzymes. This ability reinforces the potential of these compounds as candidates for treating both T. cruzi and T. brucei infections.
Nonetheless, the differences in trypanocidal potency observed between * Se * O3 and * Se * C5 suggest that secondary mechanisms, such as radical scavenging, also contribute to antiparasitic activity and may differ between species. Notably, * Se * O3 was the most potent against TbrCATL, whereas * Se * C5 was the most active against Cz, indicating that the preferred structure–activity relationship may vary depending on the specific protease target. These findings highlight the opportunity to fine-tune compound structures to selectively optimize inhibition of either protease or develop dual-target inhibitors with improved therapeutic profiles.
Finally, based on our in silico predictions of ADME properties and drug-likeness, all the compounds tested are suitable for oral administration and meet the established criteria for drug-like behavior. These predictions are supported by a pharmacokinetic study performed with * Se * C5 in our previous work? that confirmed its favorable oral bioavailability. Moreover, the in silico analysis suggests that both the open and cyclic derivatives exhibit good gastrointestinal absorption. Notably, the cyclic compounds are predicted to have a greater ability to cross the blood–brain barrier compared to the open-chain derivativesan important consideration in the context of treating CNS-infiltrated T. brucei infections.
Conclusions
We evaluated 44 chalcogen-containing compounds against T. brucei, using S-semicarbazones as a starting point and implementing an S-to-Se isosteric replacement strategy. The resulting analogues were divided into open-chain (chalcogen semicarbazones) and cyclic (selenazole) derivatives. Se-containing compounds consistently outperformed their S analogues, with the lead compound * Se * O3 showing low micromolar trypanocidal activity, nanomolar inhibition of TbrCATL, and significant antioxidant potential. These findings suggest that the dual action inhibition of TbrCATL combined with free radical scavenging may be key to the antiparasitic activity of these compounds. Although * Se * O3 meets the criteria for oral bioavailability and drug-likeness, its cytotoxicity still requires optimization. In this regard, cyclic selenazoles showed reduced toxicity while retaining activity, making them promising scaffolds. Further structural modifications, particularly on the aromatic ring, should aim to improve metabolic stability and inhibition rates. Finally, the finding of a trend in the inhibition of TbrCATL and Cz by the Se-semicarbazone- and selenazole-based derivatives might support the development of agents that target both Chagas disease and HAT.
Methods
Chemistry
The synthesis and characterization of the 44 compounds evaluated in this work were previously described by our group.? Their characterization included melting points, IR, ^1^H and ^13^C spectra. ^77^Se NMR was performed when possible. The purity (≥95%) of the compounds was determined by qNMR.
In Vitro Antitrypanosomal
Assay
Bloodstream forms of T. b. brucei Lister 427 were grown in HMI-9 modified medium? supplemented with 20% heat-inactivated fetal bovine serum (FBS) in T25 suspension cell culture flasks at 37 °C in 5% CO_2_.? Trypanosomes were maintained in exponential growth phase and passaged every 48–72 h. Growth inhibition of T. b. brucei was determined using the SYBR Green cell viability assay.? Test compounds were diluted in DMSO and added to 96-well polystyrene assay plates to give final assay concentration of 10 μM (1 μL; 0.5% total DMSO). Fresh HMI-9 medium (99 μL/well) was added to the assay plate. Parasites in exponential phase were suspended at 2 × 10^5^ parasites/mL in HMI-9 medium and added to each well (100 μL) to a total density of 2 × 10^4^ trypanosomes/well. Assay plates were incubated at 37 °C and 5% CO_2_ for 72 h, followed by addition of 50 μL/well of lysis solution (30 mM Tris, pH 7.5, 0.012% saponin, 0.12% Triton X-100 and 7.5 mM ETDA) containing 0.3 μL/mL SYBR Green I (10,000× in DMSO; Invitrogen, Carlsbad, CA). Assay plates were incubated in the dark for 1 h at room temperature. Fluorescence was measured at 485 and 535 nm excitation and emission wavelengths, respectively, using a 2104 EnVision multilabel plate reader. The viability of each well was normalized to positive and negative controls in each assay plate (pentamidine was used as the positive control). The screen was performed in technical quadruplicate. DR curves were generated for compounds inhibiting parasite growth by ≥70%. Eight-point concentration–response curves were prepared and EC_50_ values were calculated with GraphPad Prism, version 9.3 (San Diego, CA) using a sigmoidal four parameter logistic curve. EC_50_ data were generated from three independent experiments each performed in duplicate.
In Vitro Cytotoxicity Evaluation
The HEK293 and HepG2 cells were cultured in DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin-streptomycin. ?,? Cells were grown in T75 cell culture flasks maintained at 37 °C in 5% CO_2_ and subcultured when at 60–80% cell confluence. Cytotoxicity was measured using the Promega CellTiter-Glo reagent (G7572). ?,? Test compounds were diluted in DMSO and added to 96-well polystyrene assay plates to give a final assay concentration of 10 μM. Fresh medium was added to the assay plate (49 μL/well). HEK293 or HepG2 cells were suspended to 1 × 10^5^ cells/mL in DMEM and added to each well (50 μL) for a total density of 5 × 10? cells/well. Assay plates were incubated at 37 °C and 5% CO_2_ for 48 h, followed by addition of 25 μL/well of CellTiter-Glo. Luminescence was measured using a 2104 EnVision multilabel plate reader. The viability of each well was normalized to positive and negative controls in each assay plate (bortezomib was used as the positive control). CC_50_ values were calculated with GraphPad Prism, version 9.3 (San Diego, CA) using a sigmoidal four parameter logistic curve. CC_50_ data were generated from three independent experiments each performed in duplicate.
Enzyme Inhibition Assays
All experiments were performed in a 384-well black microplate, using a Synergy HTX (Biotek) plate reader at 25 °C with absorption/emission wavelengths of 360/460 nm. Assays were performed in triplicate wells and DMSO was used as a vehicle control. Enzyme activity was measured and normalized to DMSO controls, and the inhibitor E-64 (10 μM) was used as a positive control for all assays. Experiments were performed in triplicate, with at least two independent assays (n = 6 data points). IC_50_ values were determined through nonlinear regression. The data obtained were analyzed using GraphPad Prism 9.0 (GraphPad Software, La Jolla, California, US).
TbrCATL
Inhibition Assays
The screening assay was performed using 0.5 nM of TbrCATL diluted in 0.1 M sodium acetate, 1 mM dithiothreitol (DTT), and 0.01% Triton X-100, pH 5.5. TbrCATL was expressed and purified as described.? Enzyme the compound (10 μM), were incubated for 10 min at room temperature. Then the substrate, Z-Phe-Arg-amidomethylcoumarin (Z-FR-AMC; Sigma-Aldrich, C9521), was added (30 μL diluted in the same assay buffer) to yield a final concentration of 2.5 μM and the assay allowed to proceed for 10 min. The IC_50_ values of compounds inhibiting more than 85% at 10 μM and active against T. brucei (antiparasitic activity ≥ 70%) were calculated. For those compounds inhibiting enzyme activity by >85%, DR assays were performed over 15 2-fold serial dilutions starting at 0.5 or 20 μM.
hCatL Inhibition Assays
Recombinant hCatL was purchased from R&D Systems (952-CY) and activated according to the manufacturers protocol. This enzyme, hCatL, is used to test compounds’ selectivity because it is the human homologue of TbrCATL. The assay was modified from,? using 25 pM of the enzyme diluted in 40 mM sodium acetate, 5 mM DTT, 100 mM NaCl, 1 mM EDTA and 0.01% Triton X-100, pH 5.5. Enzyme and compound (10 μM), were incubated for 30 min at room temperature. Then Z-FR-AMC was added (30 μL diluted in the same assay buffer) to yield a final concentration of 25 μM and the assay allowed to proceed for 10 min. DR assays were performed over 15 2-fold serial dilutions starting at 0.5 or 20 μM.
Molecular Dynamics
The structure deposited in the Protein Data Bank (PDB) under the ID 2P7U? prepared the starting structures to run the Molecular Dynamics Simulations (MD). The ligand of the 2P7U structure was replaced by the selenium derivatives SeO1, SeO3, SeO5, SeC3 and SeC5 before running the MD simulation. Both the systems preparation and the simulations were performed in the AMBER 18 suite software. The protocol for the systems preparation and the MD simulations is detailed as follows. First, the system is neutralized by adding sodium ions and later immersed in a cubic box of 10 Å length, in each direction from the end of the protein, using TIP3P water parameters.
The force fields used to obtain topography and coordinates files were ff14SB? and GAFF.? The first step of the simulation protocol followed to run the MD simulations is a minimization of the solvent molecules position only, keeping the solute atom positions restrained, and the second stage minimizes all the atoms in the simulation cell. Heating the system is the third step, which gradually raises the temperature 0 to 300 K under a constant volume (ntp = 0) and periodic boundary conditions. In addition, Harmonic restraints of 10 kcal/mol^–1^ were applied to the solute, and the Berendsen temperature coupling scheme? was used to control and equalize the temperature. The time step was kept at 2 fs during the heating phase. Long-range electrostatic effects were modeled using the particle-mesh-Ewald method.? The Lennard-Jones interactions cutoff was set at 8 Å. An equilibration step for 100 ps with a 2 fs time step at a constant pressure and temperature of 300 K was performed prior to the production stage. The trajectory production stage kept the equilibration step conditions and was prolonged for 500 ns longer at the 1 fs time step. In addition, the selenium derivative required a previous preparation step where the parameters and charges were generated by using the antechamber module of AMBER, using the GAFF force field and AM1-BCC method for charges.?
The interaction free energy between the ligands and TbrCATL was estimated using the Molecular Mechanics Poisson–Boltzmann Surface Area (MM/PBSA) method.
Evaluation of Antioxidant
Activity
The antioxidant capacity of the selected Se-compounds was tested using the DPPH assay as described.? DPPH forms a free radical that is stable at room temperature. If the tested compounds are antioxidant, they neutralize the free radicals to donating a proton.? The compounds were tested at three different concentrations (0.06, 0.03, and 0.015 mg/mL) and we used two positive controls: ascorbic acid (ASC) and trolox (TRO). A solution of DPPH (2,2-diphenyl-1-picrylhydrazyl) in methanol (0.04 mg/mL, preserved in the dark) was prepared and 100 μL of this stock solution was added to 100 μL of tested compounds solution. The color change, from purple (radical) to yellow (reduced), was measured at 517 nm at different time points (0, 5, 15, 30, 60, 90, and 120 min). The experiment was performed three times in triplicate and the measurements were recorded on a BioTek PowerWave XS spectrophotometer (Biotek). Data were collected using BioTek Gen5Microplate reader and Imager software (Agilent, version 3.12). Data were generated from three independent experiments, each performed in triplicate (n = 9), and expressed as a percentage of inhibited DPPH (mean ± standard error of the mean (SEM)) using the following formula:
where A_control_ refers to the absorbance of the negative control and A_sample_ refers to the absorbance of the tested compounds.
In Silico Prediction of ADME and Drug-Likeness
Properties
SwissADME platform (http://www.swissadme.ch/) (accessed on 15th January 2025) was used to analyze the physicochemical and pharmacokinetic characteristics of the 44 compounds evaluated in this work. This platform is freely provided by the Swiss Institute of Bioinformatics (SIB) and provides information on bioavailability and ADME parameters (Absorption, Distribution, Metabolism and Elimination).?
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1World Health Organization Trypanosomiasis, human African (sleeping sickness). https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed 2024, August 1).
- 2DNDI Sleeping sickness: Symptoms, transmission, and current treatments for sleeping sickness. https://dndi.org/diseases/sleeping-sickness/facts/. (accessed 2024, August 1).
- 3Coetzer, J. A. W. ; Thomson, G. R. ; Tustin, R. C. Infectious diseases of livestock with special reference to Southern Africa; Oxford University Press: Cape Town, 1994.
- 4Pereira G. A.Santos L. H.Wang S. C.Martins L. C.Villela F. S.Liao W.Dessoy M. A.Dias L. C.Andricopulo A. D.Costa M. A.Benzimidazole inhibitors of the major cysteine protease of Trypanosoma brucei Future Med. Chem.2019111537155110.4155/fmc-2018-052331469332 PMC 6722484 · doi ↗ · pubmed ↗
- 5Baker N.de Koning H. P.Mäser P.Horn D.Drug resistance in African trypanosomiasis: the melarsoprol and pentamidine story Trends Parasitol.20132911011810.1016/j.pt.2012.12.00523375541 PMC 3831158 · doi ↗ · pubmed ↗
- 6Barrett M. P.Vincent I. M.Burchmore R. J.Kazibwe A. J.Matovu E.Drug resistance in human African trypanosomiasis Future Microbiol.201161037104710.2217/fmb.11.8821958143 · doi ↗ · pubmed ↗
- 7Fairlamb A. H.Horn D.Melarsoprol Resistance in African Trypanosomiasis Trends Parasitol.20183448149210.1016/j.pt.2018.04.00229705579 · doi ↗ · pubmed ↗
- 8Peerzada M. N.Gaur A.Azam A.Advances in Drug Discovery against Neglected Tropical Diseases: Human African and American Trypanosomiasis Curr. Med. Chem.2021287544758210.2174/092986732866621050411144233949927 · doi ↗ · pubmed ↗