Harnessing flagellin of Ligilactobacillus agilis as a surface display scaffold for an HIV-1 epitope
Shunya Suzuki, Gregg A. Dean, Akinobu Kajikawa

TL;DR
This study shows that flagellin from Ligilactobacillus agilis can be used to display an HIV-1 epitope, triggering immune responses and offering a new platform for mucosal vaccines.
Contribution
The first demonstration of LAB-derived flagellin as an antigen display scaffold for mucosal vaccine development.
Findings
L. agilis flagellin (FliC2) effectively displayed the HIV-1 MPER epitope on its surface.
Modified FliC2 showed improved TLR5-stimulating activity and induced both mucosal and systemic immune responses in mice.
The study establishes L. agilis flagellin as a promising platform for surface display of heterologous peptides.
Abstract
Lactic acid bacteria (LAB) are promising mucosal vaccine vectors due to their safety, immunostimulatory properties, and the availability of genetic tools for certain strains. Bacterial flagellin has attracted attention as both a versatile scaffold for antigen surface display and a potent adjuvant via Toll-like receptor 5 (TLR5) activation. Despite these advantages, LAB-derived flagellin remains largely unexplored as an antigen display platform. In this study, we demonstrate the potential of flagellin (FliC2) from Ligilactobacillus agilis, one of the few flagellated LAB species, as a scaffold for the surface display of the HIV-1 membrane-proximal external region (MPER) epitope. Recombinant L. agilis strains were engineered to express FliC2 with the MPER epitope inserted at various positions within its hypervariable domain, identifying optimal sites for effective surface display. To…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
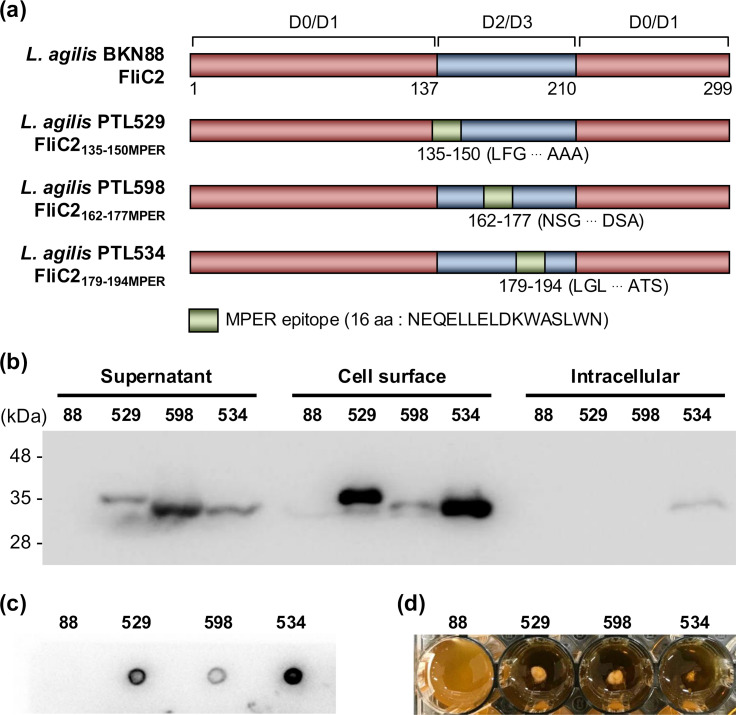 Fig 1
Fig 1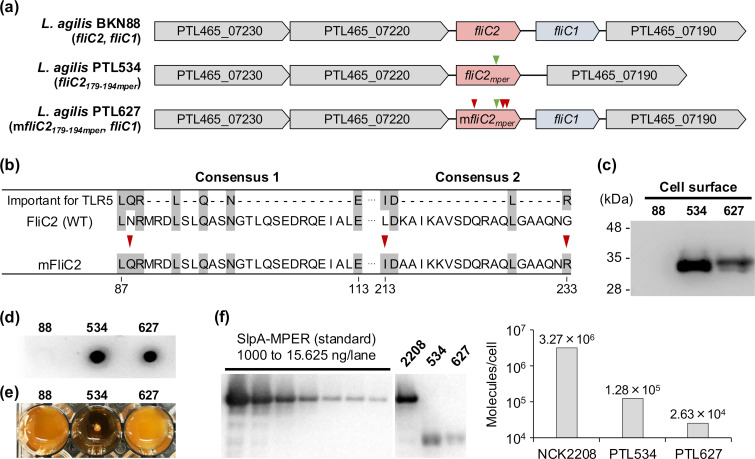 Fig 2
Fig 2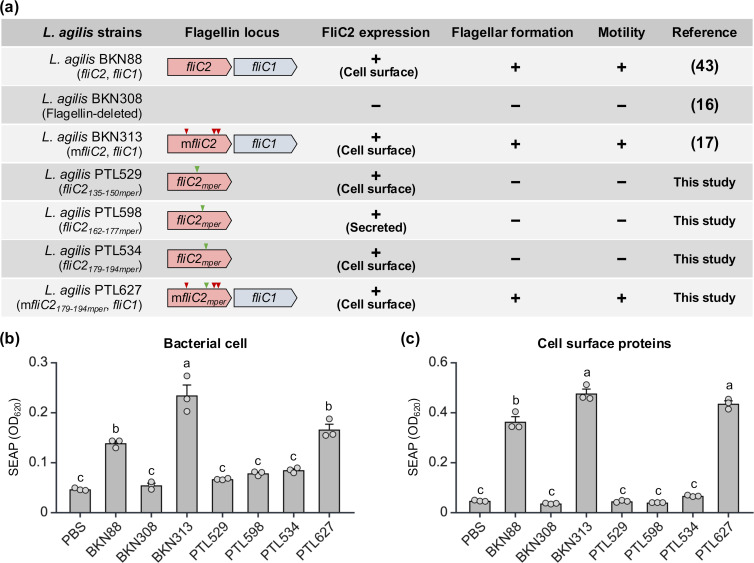 Fig 3
Fig 3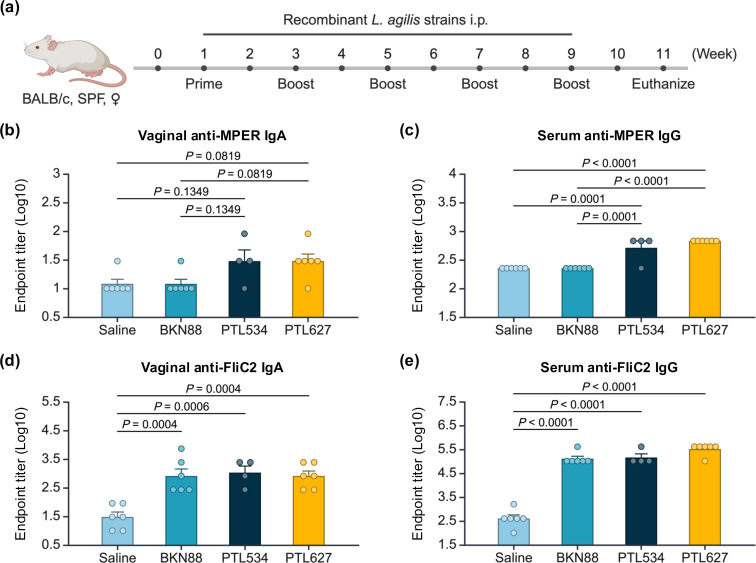 Fig 4
Fig 4| Strain or plasmid | Description and origin | Reference |
|---|---|---|
| EC101 | Cloning host for pG+host5, RepA+ | ( |
| BKN88 (WT) | Isolated from avian gut, wild type | ( |
| BKN308 | Δ | ( |
| BKN310 | Δ | ( |
| BKN313 | ( | |
| PTL529 | This study | |
| PTL534 | This study | |
| PTL598 | This study | |
| PTL627 | This study | |
| NCK2208 | ( | |
| Plasmids | ||
| pG+host5 | Replication-thermo-sensitive plasmid, Emr | ( |
| pG+host5::m | pG+host5 with mutated | ( |
| pG+host5:: | pG+host5 with | ( |
| Primer | Sequence (5′ to 3′) |
|---|---|
| pG+host5:: | |
| DOKJ797 |
|
| DOKJ798 |
|
| DOKJ799 |
|
| DOKJ800 |
|
| pG+host5:: | |
| DOKJ1062 |
|
| DOKJ1063 |
|
| DOKJ1064 |
|
| DOKJ1065 |
|
| pG+host5:: | |
| DOKJ801 |
|
| DOKJ802 |
|
| DOKJ803 |
|
| DOKJ804 |
|
| pG+host5::m | |
| DOKJ1112 |
|
| DOKJ1113 |
|
| DOKJ1114 |
|
| DOKJ1115 |
|
| Flagellin flanking region | |
| DOKJ705 |
|
| DOKJ706 |
|
| M13 primers | |
| DOKJ78 |
|
| DOKJ79 |
|
- —Tokyo University of Agriculturehttp://dx.doi.org/10.13039/501100007777
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntimicrobial Peptides and Activities · Immunotherapy and Immune Responses · Bacteriophages and microbial interactions
INTRODUCTION
Mucosal vaccines represent a promising approach to combat infectious diseases and mucosal pathobionts-related diseases due to their ability to elicit both mucosal and systemic immune responses (1). Effective mucosal immune activation requires suitable vectors to deliver antigens to mucosal sites. Lactic acid bacteria (LAB) are attractive candidates for such vectors because of their safety, immunostimulatory properties, and the availability of genetic tools for certain strains (2). LAB are generally recognized as safe (GRAS) and widely used in food production and probiotics. Certain LAB strains exhibit immunostimulatory properties and can be genetically engineered to produce heterologous proteins and peptides. These properties make LAB promising candidates for mucosal vaccine vectors (2), and LAB-based mucosal vaccines against various pathogens have been investigated (3). Notably, a recent clinical trial of an oral vaccine based on Lacticaseibacillus paracasei expressing the HPV-16 E7 antigen demonstrated safety and efficacy in treating HPV-16-positive patients with cervical intraepithelial neoplasia (4), highlighting the potential of LAB-based mucosal vaccines. Despite these advances, the efficacies of mucosal vaccines, including LAB-based vaccines, are often insufficient, and innovative strategies to enhance their efficacy are required.
Bacterial flagellin is the major structural protein of flagella, produced by various bacteria, including a few LAB species. Flagellin has the potential to play dual roles in vaccine development as both an antigen display scaffold and an adjuvant. The flagellar filament comprises approximately 20,000 flagellin subunits, enabling the extensive display of antigens on the bacterial surface (5). Moreover, flagellin is recognized by Toll-like receptor 5 (TLR5), activating innate and adaptive immune responses, thereby serving as a potential adjuvant (6–8). These structural and immunological properties suggest that flagellin could play a crucial role in enhancing mucosal vaccine efficacy. Indeed, several studies have demonstrated the efficacy of flagellin derived from Salmonella as both an antigen display scaffold and an adjuvant. For example, recombinant Salmonella expressing flagellin displaying HIV-1 V3 loop epitopes successfully induced systemic and mucosal immune responses in mice (9). Similarly, Newton et al. reported strong antibody responses when cholera toxin epitopes were inserted into Salmonella flagellin (10). While these studies highlight the potential of flagellin from pathogenic bacteria in vaccine development, LAB-derived flagellin remains largely unexplored.
Most LAB lack flagella, but Ligilactobacillus agilis is one of the few flagellated species that inhabits the gastrointestinal tracts of various animals (11). Unlike typical LAB, L. agilis exhibits motility and chemotactic abilities, which may facilitate its penetration into the mucus layer and enhance survival under harsh conditions (12, 13). Moreover, L. agilis is genetically tractable (14), allowing for the production of heterologous proteins and targeted modifications of its flagellin (15, 16). Notably, L. agilis flagellin interacts with TLR5, and targeted amino acid substitutions can enhance its immunostimulatory activity (17). These capabilities suggest that L. agilis would be a versatile vector for mucosal vaccine development.
In this study, we harnessed the flagellin (FliC2) of L. agilis as a surface display scaffold for the HIV-1 membrane-proximal external region (MPER) epitope, a critical target for broadly neutralizing antibodies (18, 19). We engineered recombinant L. agilis strains to express FliC2 with the MPER epitope inserted at targeted positions within its hypervariable domain. To enhance immunogenicity, we introduced specific mutations into the TLR5 recognition site of FliC2 to improve its adjuvant activity. Through in vitro and in vivo analyses, we evaluated the surface display of the epitope, TLR5-stimulating activity, and immunogenic potential of these recombinant L. agilis strains. Our findings provide the first evidence of utilizing LAB-derived flagellin as an antigen display scaffold, establishing L. agilis flagellin as a promising platform for mucosal vaccine development.
RESULTS AND DISCUSSION
L. agilis BKN88 possesses two active flagellin genes, fliC1 and fliC2. Our previous study showed that L. agilis forms flagella and exhibits motility with either FliC1 or FliC2 alone (16). In addition, the FliC2-expressing strain exhibited higher immunological activity via TLR5 than the FliC1-expressing strain. Based on these findings, we selected L. agilis FliC2 as the scaffold for vaccine epitope display.
Insertion of the MPER epitope into L. agilis FliC2
To determine optimal insertion sites for the MPER epitope within L. agilis FliC2, we constructed recombinant L. agilis strains expressing FliC2 variants with the MPER epitope (16 amino acid residues) inserted at different positions within its hypervariable domain. Structural analysis revealed that FliC2 consists of conserved D0/D1 domains and a hypervariable domain (Fig. 1a). Bacterial flagellins generally contain conserved D0/D1 domains and hypervariable D2/D3 domains (20), with the latter suggested to be not essential for TLR5-mediated immune activation (21). We thus replaced various regions of the hypervariable domain in L. agilis FliC2 with the MPER epitope and constructed recombinant L. agilis strains carrying each of the modified fliC2 genes into the chromosome (Fig. 1a). Western blot analysis confirmed MPER-specific bands predominantly in the cell surface fraction of L. agilis strains PTL529 (expressing FliC2_135-150MPER_) and PTL534 (expressing FliC2_179-194MPER_) (Fig. 1b). In L. agilis PTL598 (expressing FliC2_162-177MPER_), the MPER-specific band was most prominent in the culture supernatant fraction, indicating that FliC2_162-177MPER_ is primarily secreted (Fig. 1b). In dot blot analysis, the MPER-specific signal was detected in all recombinant L. agilis strains, suggesting successful surface display of the MPER epitope (Fig. 1c). The signal intensity varied among the strains, and that of L. agilis PTL529 and PTL534 was clearly discernible. These results indicate that amino acid positions 135–150 and 179–194 within L. agilis FliC2 are suitable sites for epitope insertion and surface display.
Construction and validation of recombinant Ligilactobacillus agilis strains expressing MPER-displaying flagellins. (a) Schematic representation of MPER-displaying flagellins. Domain architectures of FliC2 of L. agilis were predicted using InterPro. (b) Detection of the MPER epitope in FliC2 by Western blot using 2F5 mAb (anti-MPER monoclonal human IgG). Cell culture supernatant, cell surface, and intracellular proteins of L. agilis strains were analyzed by Western blot with 2F5 mAb. (c) Detection of the MPER epitope exposed on the cell surface of L. agilis strains by dot blot using 2F5 mAb. (d) The motility of L. agilis strains.
Although PTL529 and PTL534 produced FliC2-MPER on the cell surface, neither strain formed flagella (Fig. S1) nor exhibited motility (Fig. 1d). Bacterial flagellin is secreted via a type III secretion system (T3SS) and assembled into flagella (22). It is possible that although the modified FliC2 is transported through T3SS, it fails to assemble into functional flagella. These results suggest that while the insertion of the MPER epitope into the FliC2 hypervariable domain impairs flagellar formation and motility, the modified FliC2 still effectively displays the epitope on the bacterial surface. Hence, due to its robust FliC2-MPER production, L. agilis PTL534 was selected for further analyses to evaluate its potential as a vaccine candidate.
Construction and validation of a recombinant L. agilis expressing mutated FliC2 displaying MPER epitope
To enhance the adjuvant activity of flagellin, targeted mutations were introduced into the FliC2-MPER of L. agilis PTL534 (Fig. 2a and b). Our previous study demonstrated that substituting three specific amino acids within the TLR5 recognition site of L. agilis FliC2 significantly increases its immunostimulatory activity (17). However, an L. agilis strain carrying only the mutated fliC2 (mfliC2) could not produce mFliC2 on the cell surface. In contrast, when mfliC2 was co-expressed with wild-type fliC1, the recombinant strain successfully produced the two flagellins on the surface and was more immunogenic than the wild-type L. agilis strain. Based on these findings, we constructed L. agilis PTL627 carrying both mfliC2_179-194mper_ and fliC1 genes on its chromosome (Fig. 2a and b). Western blot analysis confirmed the production of mFliC2-MPER on the bacterial surface (Fig. 2c), and dot blot analysis demonstrated successful exposure of the MPER epitope on the surface (Fig. 2d). Further phenotypic analyses revealed that PTL627 retained flagella (Fig. S1) and exhibited motility (Fig. 2e), unlike PTL534. These results showed that L. agilis PTL627 produces MPER-displaying FliC2 with the targeted mutations while maintaining flagellar formation and motility, making it a suitable candidate for further investigation.
Construction and validation of a recombinant L. agilis expressing mutated FliC2 displaying MPER epitope. (a) Genetic map of the flagellin gene and flanking regions of L. agilis strains. The green and red arrowheads indicate epitope-replaced and fliC2-mutated sites, respectively. Locus tags are denoted in the arrows. (b) Alignment of amino acid sequences of FliC2 and mutated FliC2 (mFliC2). Conserved residues, which were reported to be important for TLR5 recognition, are highlighted in gray. The red arrowheads indicate the mutations in mFliC2. (c) Cell surface proteins of L. agilis strains were analyzed by Western blot with 2F5 mAb. (d) Detection of the MPER epitope exposed on the cell surface of L. agilis strains by dot blot using 2F5 mAb. (e) The motility of L. agilis strains. (f) Quantitative Western blot analysis of SlpA/FliC2 displaying MPER epitope on Lactobacillus acidophilus NCK2208/L. agilis strains. Left: L. acidophilus NCK2208 cells (1.75 × 106 CFU/lane), L. agilis PTL534 or PTL627 cells (3.5 × 107 CFU/lane), and the purified SlpA-MPER standards (1,000 to 15.625 ng/lane) were analyzed by Western blot with 2F5 mAb. Right: The MPER-specific signal intensity was quantified using ImageJ software, and the SlpA-MPER or FliC2-MPER molecules per cell were calculated.
Next, we estimated the amount of MPER-displaying flagellin in each bacterial cell using quantitative Western blot analysis with anti-MPER antibody (Fig. 2f; Fig. S2). The result showed that L. agilis PTL534 and PTL627 produced approximately 1.3 × 10^5^ and 2.6 × 10^4^ molecules of MPER-displaying flagellin per cell, respectively (Fig. 2f). The fivefold reduction in PTL627 compared with PTL534 may be attributed to the introduced mutations, which likely affect flagellar polymerization (20, 23, 24). Alternatively, the additional expression of fliC1 might have affected flagellin production. Our analysis also estimated that Lactobacillus acidophilus NCK2208 (25), which produces MPER-displaying surface layer protein (SlpA-MPER), produced approximately 3.3 × 10^6^ molecules of SlpA-MPER per cell (Fig. 2f). Compared with L. acidophilus NCK2208, L. agilis PTL534 and PTL627 produced approximately 25- and 124-fold fewer MPER-displaying protein molecules, respectively. Nevertheless, the capacity to display epitopes of L. agilis flagellin is still comparable to that of other surface display systems in LAB, supporting its feasibility for vaccine development. For example, approximately 1.4 × 10^3^ to 3.9 × 10^3^ molecules per cell of tetanus toxin fragment C (TTFC) fused to a PrtP-derived LPXTG anchor have been displayed on the surface of Lacticaseibacillus casei (26). Other surface display systems, such as those based on LysM- and SLP-derived anchoring domains, have been reported to display approximately 4.5 × 10^3^ to 1.0 × 10^6^ and 1.1 × 10^6^ to 1.0 × 10^7^ molecules per cell, respectively (27–31). Collectively, these findings suggest that L. agilis FliC2 serves as an effective antigen display scaffold, while its expression levels vary depending on the presence of the targeted mutations and/or co-expression of FliC1.
TLR5-mediated immunological activity of recombinant L. agilis strains
The TLR5-stimulating activity of a series of L. agilis flagellin mutants (Fig. 3a) and their cell surface proteins was evaluated using a TLR5-reporter gene assay. As shown in Fig. 3b, BKN313 (expressing mFliC2 and FliC1) showed the highest TLR5 activation, while PTL529 (expressing FliC2_135-150MPER_), PTL598 (expressing FliC2_162-177MPER_), and PTL534 (expressing FliC2_179-194MPER_) exhibited little or no activity, similar to the flagellin-deficient strain BKN308. The activity of the wild-type strain BKN88 (expressing FliC2 and FliC1) and PTL627 (expressing mFliC2_179-194MPER_ and FliC1) was intermediate. A comparable pattern was observed in the TLR5-stimulating activity of the cell surface protein fractions, although the activity of PTL627 exhibited higher activity than that of BKN88 (Fig. 3c). Despite the production of FliC2-MPER in L. agilis PTL529, PTL598, and PTL534 (Fig. 1c and d), their TLR5-stimulating activity was as low as that of the flagellin-deleted strain BKN308. In contrast, our previous study showed that L. agilis expressing only FliC2 exhibited significantly higher TLR5-stimulating activity than BKN308 (16). These results indicate that the reduced activity of the FliC2-MPER-expressing strains is likely due to replacing the FliC2 hypervariable domain with the MPER epitope, rather than the absence of FliC1. Although several studies have demonstrated that the hypervariable domain is not essential for TLR5 activation (21, 32–35), Biedma et al. reported that deletion of this domain in Salmonella flagellin altered the secondary structure of the TLR5 recognition site, thereby reducing its activity via human TLR5 (hTLR5) but not mouse TLR5 (mTLR5) (36). Since our assays used HEK293 cells expressing hTLR5, it is possible that the MPER insertion affected the ability of FliC2 to activate hTLR5.
Immunological activity of recombinant L. agilis strains mediated via TLR5. (a) Genotypic and phenotypic characteristics of recombinant L. agilis strains. Partial genetic map of the flagellin gene of L. agilis strains is shown. The green and red arrowheads indicate epitope-replaced and fliC2 mutated sites, respectively. FliC2 expression was detected by either Western blot with 2F5 mAb or SDS-PAGE and CBB staining. Flagellar filaments were stained with FLAGELLA STAIN (Hardy Diagnostics) and observed using optical microscopy. The motility of L. agilis strains was observed using a semisolid MRS medium. (b and c) TLR5-reporter gene assay with whole L. agilis cells from different strains (b) and their cell surface proteins (c). Bacterial cells (1.0 × 107 CFU/well) or cell surface proteins extracted from 1.0 × 107 CFU of bacterial cells were added to HEK-Blue-hTLR5 cells (2.5 × 104 cells/well), and released secreted alkaline phosphatase (SEAP) was measured. Values are mean plus standard error (n = 3). Statistical significance was determined by one-way ANOVA with Tukey multiple comparisons test. Different letters represent statistical significance.
As expected, PTL627, which harbors both mfliC2_179-194mper_ and fliC1, exhibited enhanced TLR5-stimulating activity, surpassing that of BKN88 (Fig. 3c). This increased activity may be attributed to the triple mutations introduced in the TLR5 recognition site of FliC2, as well as the additional expression of FliC1. Previous studies have shown that L. agilis expressing only FliC1 shows significantly lower TLR5 activation than that of BKN88 (16), supporting the idea that the mutations in FliC2 contributed to the enhanced TLR5 activation. As TLR5 activation by flagellin plays a critical role in promoting both innate and adaptive immune responses, L. agilis PTL627 may have potential as a vaccine candidate.
Immunological potentials of recombinant L. agilis strains expressing MPER-displaying FliC2
To evaluate the immunological potential of MPER-displaying L. agilis, mice were immunized intraperitoneally with either L. agilis BKN88, PTL534, or PTL627, and the production of MPER- and FliC2-specific antibodies was measured (Fig. 4a). MPER-specific IgA titers in vaginal washes tended to increase in the PTL534 and PTL627 immunized groups compared with the BKN88 and saline groups, suggesting their ability to induce mucosal immune responses (Fig. 4b). In addition, immunization with PTL534 and PTL627 significantly elevated MPER-specific serum IgG titers, indicating a robust systemic antibody response against the epitope (Fig. 4c). These results demonstrate that the MPER epitope displayed on L. agilis flagellin can elicit both mucosal and systemic immune responses.
Immunological potentials of recombinant L. agilis strains expressing MPER-displaying flagellins. (a) Experimental schema. Mice were intraperitoneally immunized with either L. agilis BKN88, L. agilis PTL534, L. agilis PTL627, or sterile saline at 2 week intervals for a total of five immunizations (n = 4 to 6 mice/group). Two weeks after the last immunization, serum and vaginal washes were collected for quantification of antibody titers. (b and c) Endpoint titers of MPER-specific vaginal IgA (b) and serum IgG (c). (d and e) Endpoint titers of FliC2-specific vaginal IgA (d) and serum IgG (e). Values are mean plus standard error, and each dot represents an individual mouse. Statistical significance was determined by one-way ANOVA with Tukey multiple comparisons test, and P-values are shown.
Endpoint titers of FliC2-specific IgA and IgG were comparable between all L. agilis-immunized groups and were significantly higher than those in the saline-immunized group (Fig. 4d and e). Interestingly, despite the differences in hTLR5-stimulating activity between PTL534 and PTL627, both strains elicited similar levels of MPER- and FliC2-specific antibodies. This discrepancy could be attributed to the approximately fivefold difference in FliC2-MPER production between PTL534 and PTL627, which may have influenced immune activation. Additionally, differences in flagellin recognition between human and mouse TLR5 could also account for the variation in immune activation. Previous studies have shown that mTLR5 is more tolerant of mutations and deletions in flagellin than hTLR5, which is more sensitive to structural changes of flagellin (36, 37). Thus, while L. agilis PTL534 exhibited minimal hTLR5-stimulating activity in vitro, it might still activate mTLR5, leading to comparable immune responses to PTL627 in vivo. However, there is no direct evidence to support this, and further studies are needed.
Collectively, these findings demonstrate that L. agilis flagellin is an effective antigen display platform, capable of eliciting both mucosal and systemic immune responses.
Conclusion
This study demonstrates that L. agilis FliC2 can serve as both an antigen display scaffold and an immune activator. By inserting the MPER epitope into the hypervariable domain of FliC2, we successfully engineered L. agilis strains that effectively expose the epitope on their surface. Furthermore, targeted mutations in the TLR5 recognition site enhanced its immunostimulatory activity. Immunization with MPER-displaying L. agilis induced both mucosal and systemic MPER-specific antibody responses, underscoring its potential as a mucosal vaccine platform. This study provides the first evidence that L. agilis flagellin can function as an antigen delivery vector, offering a promising foundation for future mucosal vaccine development. However, since the immune responses in this study were evaluated following intraperitoneal immunization, further research is necessary to determine whether MPER-displaying L. agilis can elicit immune responses when administered through oral or intranasal routes. In addition, the roles of bacterial motility and TLR5-stimulating activity in antigen-specific immune induction remain to be elucidated. Despite the differences in motility and TLR5-stimulating activity between PTL534 and PTL627, both strains induced comparable antigen-specific immune responses. Further investigation into the impact of these factors—for example, through comparisons between PTL627 and an L. agilis mutant expressing FliC2-MPER and FliC1—could provide valuable insights for the rational design of more effective mucosal vaccines. Finally, while MPER-specific antibodies were successfully induced, their ability to neutralize HIV-1 remains to be determined. Future studies should investigate whether immunization with recombinant L. agilis confers protection against HIV-1 or other pathogens in relevant infection models. Such investigations will provide critical insights into the clinical applicability of this vaccine platform.
MATERIALS AND METHODS
Bacterial strains and growth conditions
The bacterial strains and plasmids used in this study are listed in Table 1. L. agilis BKN88, its derivative strains, and L. acidophilus were anaerobically propagated in MRS broth or agar (Difco, BD, USA) with or without 5 µg/mL of erythromycin at 37°C. E. coli EC101 and derivative strains were aerobically grown in Brain Heart Infusion (Difco, BD, USA) broth or agar with or without 200 µg/mL of erythromycin and 40 µg/mL of kanamycin at 37°C.
Construction of recombinant L. agilis strains
Four kinds of recombinant L. agilis strains, L. agilis PTL529, PTL534, PTL598, and PTL627, were constructed by inserting fliC2_135-150mper_, fliC2_179-194mper_, fliC2_162-177mper_, or mfliC2_179-194mper_, and fliC1 into the chromosome of the flagellin-deleted BKN308 strain of L. agilis, respectively. The plasmids and primers used for constructing the recombinant L. agilis strains are listed in [Tables 1 and 2](#T1 T2). The plasmids for inserting the modified flagellin gene were constructed from pG^+^host5::fliC2 (16) or pG^+^host5::mfliC2, fliC1 (17) using the in-Fusion HD Cloning Kit (Takara-bio, Shiga, Japan). The linearized plasmids were amplified by inverse PCR using primer pairs DOKJ797 and DOKJ798, DOKJ801 and DOKJ802, DOKJ1062 and DOKJ1063, or DOKJ1112 and DOKJ1113 with pG^+^host5::fliC2 or pG^+^host5::mfliC2, fliC1 as the template. MPER-encoding sequences were amplified by PCR using primer pairs DOKJ799 and DOKJ800, DOKJ803 and DOKJ804, DOKJ1064 and DOKJ1065, or DOKJ1114 and DOKJ1115. Chromosomal DNA of L. acidophilus NCK2208 was used as template. The linearized plasmid and the amplified MPER fragment were ligated using the In-Fusion HD Cloning Kit (Takara-bio, Shiga, Japan) and introduced into E. coli EC101. The transformants harboring plasmids with the desired insert were selected by colony PCR and Sanger sequencing using the M13 primers, DOKJ78 and DOKJ79. The constructed plasmids were introduced into L. agilis BKN308 by electroporation, and the modified flagellin gene was then inserted into the chromosome of BKN308 by double-crossing over, as described previously (16). The insertions of fliC2_135-150mper_, fliC2_179-194mper_, fliC2_162-177mper_, or mfliC2_179-194mper_, and fliC1 genes in the constructed L. agilis strains, PTL529, PTL534, PTL598, and PTL627, were confirmed by PCR and Sanger sequencing using DOKJ705 and DOKJ706 primers.
SDS-PAGE and Western blot
L. agilis strains were cultured overnight in MRS broth, and the overnight culture was centrifuged at 10,000 × g for 10 min at 4°C. The culture supernatant was precipitated with trichloroacetic acid (TCA), and the precipitated proteins were washed with acetone, followed by resuspension in 0.05 vol of 8 M urea. This suspension was used as cell culture supernatants. Cell surface proteins were extracted using 8 M urea. The bacterial cell pellet was washed twice with phosphate-buffered saline (PBS) and suspended in 0.05 vol of 8 M urea. Following vigorous homogenization, the bacterial cell suspension in 8 M urea was centrifuged at 10,000 × g for 10 min at 4°C, and the supernatant was collected as cell surface proteins. Intracellular proteins were prepared from the 8 M urea-treated cells with bead beating. The 8 M urea-treated cells were resuspended in 0.05 vol of 8 M urea, followed by bead beating with a FastPrep Instrument (MP Biomedicals, Santa Ana, CA, USA). Cellular debris was removed by centrifugation, and cell extracts were subsequently obtained as intracellular proteins. These samples were mixed with an equal volume of 2× Laemmli buffer (BIO-RAD, Hercules, CA, USA) containing 5% beta-mercaptoethanol and boiled for 5 min. The proteins were separated onto 5%–20% SDS-PAGE gels (c-PAGEL HR, ATTO, Tokyo, Japan), and the gels were transferred onto a PVDF membrane (Millipore, Burlington, MA, USA) for Western blotting. The membranes were blocked with 1% bovine serum albumin (BSA) in PBS containing 0.05% Tween 20 (PBST) for 1 h at room temperature with agitation and then incubated with 1% BSA in PBST containing the primary antibody (anti-HIV-1 gp41 monoclonal IgG, 2F5, which was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH [Dr. Hermann Katinger]) for 1 h at room temperature with agitation. The membrane was washed thrice with PBST and incubated with 1% BSA in PBST containing horseradish-peroxidase (HRP)-conjugated anti-human IgG (Cell Signaling Technology Inc, Danvers, MA) for 1 h at room temperature with agitation. After the membrane was washed five times with PBST, MPER-specific signals were developed with Luminata Forte Western HRP Substrate (Millipore, USA) and visualized by ChemiDoc XRS + imaging system (Bio-Rad).
Dot blot
L. agilis strains were cultured overnight in MRS broth, and the cells were harvested by centrifugation at 6,000 × g for 5 min at 4°C. The cell pellet was washed thrice with PBS and suspended in methanol. The whole-cell suspensions in methanol were then spotted on the PVDF membrane (Millipore, USA), and dried at room temperature for 20 min. The membrane was incubated with 1% BSA in PBS containing the primary antibody (anti-HIV-1 gp41 monoclonal IgG, 2F5) for 30 min at room temperature with agitation. The membrane was washed twice with PBS and incubated with 1% BSA in PBS containing the secondary antibody (HRP-conjugated anti-human IgG) for 30 min at room temperature with agitation. After the membrane was washed thrice with PBS, MPER-specific signals were developed with Luminata Forte Western HRP Substrate (Millipore, USA) and visualized by ChemiDoc XRS + imaging system (Bio-Rad).
Isolation of flagellar filaments
The flagellar filaments were isolated as previously described (16). Briefly, bacterial cells in the mid-exponential phase were harvested and suspended in distilled water. The flagellar filaments were removed from the cells by homogenization using a FastPrep Instrument (MP Biomedicals, Santa Ana, CA, USA) at a speed setting of 6.5 m/s for 2 min. The cells were pelleted by centrifugation at 12,000 × g for 15 min at 4°C, and the supernatant containing the flagella was subsequently fractionated by ultracentrifugation at 100,000 × g for 1 h at 4°C. The precipitated flagella were suspended in a small amount of distilled water and stored at −20°C until use.
Motility test and flagellar staining
The motility of L. agilis strains was observed using a semisolid MRS medium. L. agilis cells were inoculated into semisolid MRS medium containing 0.15% (w/v) agar and incubated at 37°C for 1 day. The cells that had migrated after growth in the semisolid MRS medium were observed. Bacterial motility in the mid-exponential phase was also observed using a BZ-X710 microscope (KEYENCE, Osaka, Japan). The flagellar filaments of L. agilis strains were stained with FLAGELLA STAIN (Hardy Diagnostics) according to the manufacturer’s protocol. The stained cells were subsequently visualized using a BZ-X710 microscope (KEYENCE, Osaka, Japan).
In silico analysis
The GenBank accession number of L. agilis FliC2 is WP_172576007.1. Domain architectures of L. agilis FliC2 were analyzed with InterPro (41).
Quantitative Western blot analysis
Extraction and purification of SlpA-MPER from L. acidophilus NCK2208 were performed with LiCl as described previously (42). The protein concentrations were determined using a Protein Assay Rapid kit wako II (Wako, Japan) according to the manufacturer’s protocol. The purified SlpA-MPER was used as a standard for quantitative western blotting. Bacterial cells in the exponential phase were harvested via centrifugation at 6,000 × g for 5 min at 4°C and gently washed twice with cold PBS. The cell pellet was suspended in PBS, then diluted and plated on MRS plates for enumeration. After centrifugation, the cell pellet was stored at −20°C until use. The bacterial cell pellet was suspended in 8 M urea and vigorously homogenized. The bacterial cell suspension and 2-fold serial dilution of the purified SlpA-MPER were mixed with an equal volume of 2 × Laemmli buffer (BIO-RAD, Hercules, CA, USA) containing 5% beta-mercaptoethanol and boiled for 5 min. L. acidophilus NCK2208 cells (1.75 × 10^6^ CFU/lane), L. agilis PTL534 or PTL627 cells (3.5 × 10^7^ CFU/lane), and the SlpA-MPER standards (1000 to 15.625 ng/lane) were loaded onto 5-20% SDS-PAGE gels (c-PAGEL HR, ATTO, Tokyo, Japan), and MPER-specific signals were detected by western blot with 2F5 mAb as described above. Western blot images were processed, and MPER-specific signal intensity was quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA), according to a previous study (43, 44). Subsequently, the SlpA-MPER or FliC2-MPER molecules per cell (CFU) were calculated from the acquired data. At least two independent experiments were performed.
TLR5 reporter gene assay
Immunological activity via TLR5 was analyzed using HEK‐Blue‐hTLR5 cells (InvivoGen) according to the manufacturer’s instructions. Briefly, HEK-Blue-hTLR5 cells were maintained in Dulbecco’s modified Eagle medium (DMEM; Wako) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (BD), 2 mM L-glutamine, 100 U/mL of penicillin (Gibco), 100 mg/mL of streptomycin (Gibco), 100 mg/mL of normocin (InvivoGen), 30 µg/mL of blasticidin (InvivoGen), and 100 µg/mL of zeocin (InvivoGen) in 5% CO2 at 37°C. Semi-confluent cultures of HEK-Blue-hTLR5 cells were harvested with cell scrapers. The cells were diluted with HEK-Blue Detection medium (InvivoGen) and then seeded to 96-well flat-bottom microplate wells (Thermo Fisher Scientific) at a concentration of 2.5 × 10^4^ cells/well. Bacteria cells from an overnight culture were harvested by centrifugation at 6,000 × g for 5 min at 4°C and washed three times with PBS, followed by resuspension in PBS. Cell surface proteins from the L. agilis cells were extracted by using 8 M urea. Bacterial cells were suspended in 8 M urea and vigorously homogenized. Following centrifugation at 10,000 × g for 10 min at 4°C, the supernatant was collected and diluted with PBS. Bacterial cells (1.0 × 10^7^ CFU/well) or cell surface proteins extracted from 1.0 × 10^7^ CFU of bacterial cells were added to each well and incubated in 5% CO_2_ at 37°C for 16 h. Secreted alkaline phosphatase (SEAP) production levels were determined by measuring the optical density at 620 nm (OD_620_) using an SH-1000 Lab Microplate Reader (Corona Electric).
Immunization of mice
Female BALB/c mice were obtained from Crea Japan. Female BALB/c mice (10 weeks old) were intraperitoneally immunized with either L. agilis BKN88, L. agilis PTL534, L. agilis PTL627, or sterile saline. The L. agilis strains were cultured overnight in MRS broth, and the bacterial cells were collected by centrifugation at 6,000 × g for 5 min at 4°C. The cells were then washed thrice with PBS and suspended in sterile saline. Mice (4 to 6 mice/group) were immunized with 5.0 × 10^8^ CFU (for prime immunization) or 1.0 × 10^8^ CFU (for boost immunization) of the L. agilis strains, or saline at 2 week intervals for a total of 5 immunizations. Mice were euthanized 2 weeks after the last immunization, and blood was collected by cardiac puncture. The serum was separated by centrifugation at 3,000 × g for 10 min at 4°C. Vaginal lavage was collected by washing the vagina with PBS containing 0.05% Tween 20 using a pipet. Insoluble debris was removed by centrifugation at 10,000 × g for 10 min at 4°C. These samples were stored at −20°C until use.
Enzyme-linked immunosorbent assay (ELISA) for quantification of antibody titers
Ninety-six-well flat-bottom microplate wells (Thermo Fisher Scientific) were coated with either 1 µg/mL of synthetic MPER peptide (Bio-Synthesis Inc, Lewisville, TX) or 5 µg/mL of purified FliC2 isolated from L. agilis BKN310 cells in carbonate buffer (15 mM Na_2_CO_3_ and 35 mM NaHCO_3_; pH 9.6) overnight at 4°C. After washing three times with PBST, the wells were blocked with 1% BSA in PBS for 1 h at room temperature and washed three times with PBST. Samples were serially diluted with 1% BSA in PBST and added to each well, followed by incubation for 2 h at room temperature. Following washing five times with PBST, the wells were subsequently incubated with either HRP-conjugated anti-mouse IgG (A2304, SIGMA) for the serum samples or HRP-conjugated anti-mouse IgA (ab97235, abcam) for the vaginal samples. After incubation for 1 h at room temperature, the wells were washed seven times with PBST, and the color was developed by incubating with 3,3′,5,5′-tetramethylbenzidine (TMB) for 30 min at room temperature. The reaction was stopped with 2N H_2_SO_4_, and the absorbance was measured at 450–570 nm using an SH-1000 Lab Microplate Reader (Corona Electric). The mean value plus 3 standard deviations (SD) of the samples from the saline-immunized group was applied as the cut-off value to determine the endpoint titer.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lavelle EC, Ward RW. 2022. Mucosal vaccines - fortifying the frontiers. Nat Rev Immunol 22:236–250. doi:10.1038/s 41577-021-00583-234312520 PMC 8312369 · doi ↗ · pubmed ↗
- 2Wells JM, Mercenier A. 2008. Mucosal delivery of therapeutic and prophylactic molecules using lactic acid bacteria. Nat Rev Microbiol 6:349–362. doi:10.1038/nrmicro 184018345021 PMC 7096801 · doi ↗ · pubmed ↗
- 3Le Cureux JS, Dean GA. 2018. Lactobacillus mucosal vaccine vectors: immune responses against bacterial and viral antigens. m Sphere 3:e 00061-18. doi:10.1128/m Sphere.00061-1829769376 PMC 5956152 · doi ↗ · pubmed ↗
- 4Kawana K, Kobayashi O, Ikeda Y, Yahata H, Iwata T, Satoh T, Akiyama A, Maeda D, Hori-Hirose Y, Uemura Y, Nakayama-Hosoya K, Katoh K, Katoh Y, Nakajima T, Taguchi A, Komatsu A, Asai-Sato M, Tomita N, Kato K, Aoki D, Igimi S, Kawana-Tachikawa A, Schust DJ. 2023. Phase I and II randomized clinical trial of an oral therapeutic vaccine targeting human papillomavirus for treatment of cervical intraepithelial neoplasia 2 and 3. JNCI Cancer Spectr 7. doi:10.1093/jncics/pkad 101PMC 1074857838001029 · doi ↗ · pubmed ↗
- 5Westerlund-Wikström B. 2000. Peptide display on bacterial flagella: principles and applications. Int J Med Microbiol 290:223–230. doi:10.1016/S 1438-4221(00)80119-810959724 · doi ↗ · pubmed ↗
- 6Cui B, Liu X, Fang Y, Zhou P, Zhang Y, Wang Y. 2018. Flagellin as a vaccine adjuvant. Expert Rev Vaccines 17:335–349. doi:10.1080/14760584.2018.145744329580106 · doi ↗ · pubmed ↗
- 7Mizel SB, Bates JT. 2010. Flagellin as an adjuvant: cellular mechanisms and potential. J Immunol 185:5677–5682. doi:10.4049/jimmunol.100215621048152 PMC 3756556 · doi ↗ · pubmed ↗
- 8Hajam IA, Dar PA, Shahnawaz I, Jaume JC, Lee JH. 2017. Bacterial flagellin-a potent immunomodulatory agent. Exp Mol Med 49:e 373. doi:10.1038/emm.2017.17228860663 PMC 5628280 · doi ↗ · pubmed ↗