CompMap: an allele-specific expression read counter based on competitive mapping
Santiago Sanchez-Ramirez, Asher Cutter

TL;DR
CompMap is a new method for analyzing gene expression differences between species or populations by accurately counting allele-specific reads.
Contribution
CompMap introduces a streamlined, self-contained approach to allele-specific expression analysis with reduced mapping bias and information loss.
Findings
CompMap accurately sorts and counts reads in homologous regions using parental alignments.
Ambiguous alignments are resolved using either proportional or statistical methods.
Simulations show CompMap has low error rates in detecting regulatory divergence.
Abstract
Gene regulatory changes acting cis and trans to a gene can be inferred with allele-specific expression (ASE) transcriptomes from interspecies and inter-population hybrids and their parents. Problems of mapping bias and excessive information loss, however, can arise unintentionally from cumbersome analysis pipelines. We introduce CompMap, a self-contained method in Python that generates allele-specific expression counts from genotype-specific alignments. CompMap sorts and counts reads, not just SNPs, by comparing read-mapping statistics to parental alignments within homologous regions. Ambiguous alignments resolve proportionally to allele-specific counts or statistically using a binomial distribution. Simulations with CompMap show low error rates in assessing regulatory divergence.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
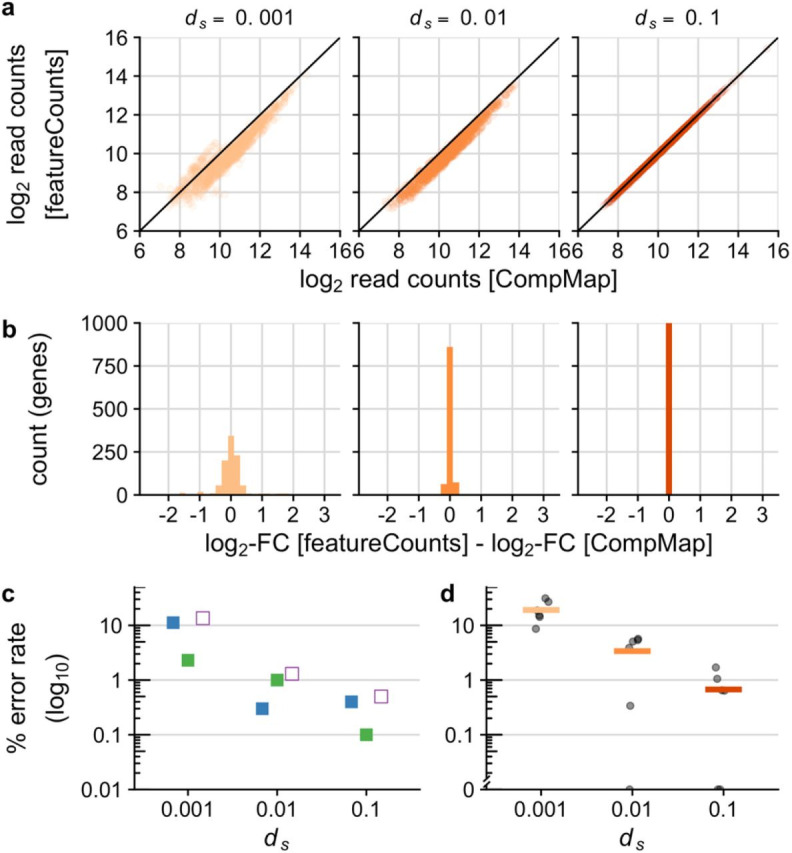 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Gene expression and cancer classification · Genomics and Chromatin Dynamics
Description
Geneticists' long-standing quest to understand functional variation within genomes has led to the appreciation that changes in regulatory regions are crucial in fine-tuning developmental controls of gene expression to influence phenotypes, in addition to the effects of structural changes in protein-coding genes (The ENCODE Project Consortium, 2012; Signor and Nuzhdin, 2018). Non-coding mutations to cis-regulatory elements that are localized close to a gene and transregulatory factors that are encoded at a distant location in the genome provide the genetic variation for expression of a focal gene, which, in turn, can drive adaptive divergence between populations and between species (Jones et al., 2012; Wray, 2007). However, identifying genetic variants with cis-acting or trans-acting regulatory effects presents a serious challenge, as it is difficult to disentangle without explicit allele-specific expression (ASE) information (Signor and Nuzhdin, 2018).
Current ASE approaches include lengthy pipelines with multiple dependencies aimed at within-species datasets (Rozowsky et al., 2011), tissue-specificity (Pirinen et al., 2015), and cross-population samples (Fan et al., 2020), usually requiring reads to be mapped to a single genotype-specific reference, followed by variant-calling and phasing. That said, the field has been growing rapidly in the last decade, resulting in highly accurate tools (e.g., van de Gejin et al., 2015; Raghupathy et al., 2018; Adduri and Kim, 2024). Here, we present CompMap, a simple, self-contained, variant-calling-free Python tool for counting reads in ASE datasets. The motivation for developing CompMap stems from the need for a tool that circumvents complex bioinformatic pipelines while avoiding the limitations of variant-calling and generates read counts compatible with other downstream read-counting software.
Methods
Software
The Python code with examples and installation instructions is available on the GitHub repository in "Extended Data". In brief, CompMap parses two F1 hybrid BAM files with RNA-seq reads mapped to each parental genome simultaneously and compares the alignment quality of each read against both references. Allele-specific reads are then "competitively" assigned, based on their relative mapping scores, and counted. CompMap is fully implemented in Python, relying on the pysam API library to read and parse BAM files and numpy (e.g., for binomial distribution corrections). It requires the following basic input files:
BAM files with F1 hybrid (offspring) RNA-seq data mapped to each parental genotypeBED files with genomic coordinates of the genes of interest, one per parental genotype
Preferably, BAM files are sorted and indexed. The 4th column of BED files should include the gene name and match for homologous regions between parental genomes (i.e., same name for both BED files). The -h or --help argument will print descriptions and other information to the screen. The two most important arguments are --AS_tag and --NM_tag, which indicate labels for the alignment score and number of mismatches tags, respectively. By default, CompMap will use tags from the STAR aligner (Dobin et al., 2012). The user can specify read-tag labels of different aligners, such as BWA (Li and Durbin, 2010) and Bowtie2 (Langmead and Salzberg, 2012). The --star argument will apply the NH tag when looking for multiple read matches, which may improve speed. Finally, users can specify the --binom tag to assign ambiguous read counts probabilistically to either one of the parental alleles. The binomial probability p describes the fraction of non-ambiguous reads with better matches to one parental genome for the corresponding gene (1- p better matching the other parental genome), and size defines the total number of ambiguously matching reads. The default behavior deterministically allocates reads with proportions p and 1- p without using binomial sampling.
We validated the read-counting performance of CompMap by comparing it to two commonly used differential expression tools: featureCounts (Liao et al., 2014) and HTseq-counts (Anders et al., 2015). Our results replicate the results of featureCounts and therefore recommend using this tool for read-counting parental RNA-seq data which can be used in combination with CompMap.
Validation on simulated data
Gene length and sequence differences between alleles can potentially impact the power to detect and quantify ASE with accuracy. To validate our approach, we simulated protein-coding sequence datasets of homologous alleles with synonymous site divergence. We neglected divergence at nonsynonymous sites because of their rarity in biological data of close relatives and to avoid assumptions about the strength and direction of selection. The presence of nonsynonymous differences in real data will make our power analysis conservative with respect to detecting and quantifying ASE.
The simulation procedure first drew 1000 random protein lengths from a Gamma distribution (scale=1000, shape=1.35; minimum length 300 aa). Non-stop codons were picked at random to form a "transcript", all of which started with the Methionine coding "ATG" and ended with any of the three stop codons. Synonymous substitutions were imposed to create a divergent version of every "transcript": each 4-fold, 3-fold, and 2-fold degenerate codon was allowed to mutate to a synonymous codon, selecting alternate codons with equal probability. Synonymous divergence used one of three rates: (1) high * d s * =0.1 substitutions/site (exemplifying interspecific variation; e.g., between species of Caenorhabditis ), (2) moderate * d s * =0.01 substitutions/site (exemplifying variation between divergent populations), and (3) low * d s * =0.001 substitutions/site (exemplifying intraspecific variation; e.g. humans). CompMap's GitHub repository contains the code for this simulation procedure in "Extended Data".
For each "allelic" transcript sequence, we then simulated RNA-seq reads using the R package polyester (Frazee et al., 2015). Fold-changes in expression were randomly sampled from an exponential distribution with sign (up- or down-regulation) assigned randomly. These generated data defined "true" ASE counts for comparison to estimates derived from CompMap. Similarly, we generated RNA-seq datasets representing expression differences in each homozygous parent. Fold-change values between "alleles" and "parents" were drawn from independent distributions.
To validate ASE counts from CompMap, we combined allele-specific reads into a single FASTA file for each replicate and mapped them to each transcript reference using BWA-MEM (Li and Durbin, 2010). We then generated BED files with coordinates for each transcript. The resulting BAM and BED files were fed to CompMap to perform ASE counts using the --NM_tag with NM, providing the BWA-specific read tag for number of mismatches.
Standard read counts for "parental" and true "allele" read data were performed with featureCounts (Liao et al., 2014). Raw read counts were then analyzed in R, and differential expression analyses were conducted with DESeq2 (Love et al., 2014). We followed McManus et al. (2010) to statistically assign genes to different regulatory divergence categories (e.g., cis -only, trans -only, cis-trans compensatory, *cis * x trans ).
Statistical Analyses
CompMap recovered high accuracy in allele-specific counts for all three simulated protein-coding datasets that spanned a 100-fold range of divergence (Figure 1a). Count accuracy was highest for genes with high divergence between alleles ( * d s * =0.1). In the more challenging case of low divergence ( * d s * =0.001), CompMap slightly overestimated the number of reads for a given allele (by a factor of approximately 1.75), being most pronounced among genes with a lower magnitude of expression, as expected. However, this effect does not strongly perturb analyses of differential expression (Figure 1b). High-divergence alleles also show the lowest variance in the ASE log 2 -fold-difference between simulated allele-specific reads counted by featureCounts and CompMap (Figure 1b). Despite the low divergence dataset yielding the widest variability in estimated differential expression, the mean centered close to zero indicates little bias (Figure 1b).
Error rates to inferring differential expression between alleles declined approximately 10-fold with increasing divergence (Figure 1c). The power of CompMap to accurately classify genes into different regulatory divergence categories is greatest with high divergence between alleles (<2% false negative rates for the * d s * =0.1 dataset; <6% for intermediate * d s * =0.01) (Figure 1d). As expected, the highest inaccuracy occurred with low divergence ( * d s * =0.001 false negative rates ~20%).
Consequently, ASE reliability will be greatest for biological studies involving alleles with high sequence divergence, which will be challenging with neutral allele divergence ~0.1% (e.g., * d s * =0.001), as within humans (Perry et al., 2012) and Caenorhabditis elegans (Andersen et al., 2012). Moreover, although our specific use case involved high-quality reference genomes from C. briggsae and C. nigoni (Sánchez-Ramírez et al., 2021), we anticipate that differences in reference genome quality might also lead to mapping biases and other potential issues. Essential for such cases is a simulation framework for validation, as implemented in CompMap, to assess the power to quantify ASE.
CompMap's competitive read-mapping approach is sensitive and accurate, given sufficiently dense sequence differences between alleles. With CompMap, we 1) introduced a framework to reliably assess the power of recovering per-gene ASE read counts; 2) developed R code for simulation testing of coding sequence divergence; and 3) showed the best ASE inference with ≥1% allelic synonymous divergence. CompMap is ideal for RNA-seq datasets derived from interspecies hybrids (Sánchez-Ramírez et al., 2021), as well as within-species analysis of systems with high genetic diversity, including *Drosophila * and *Caenorhabditis * (Cutter et al., 2013).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adduri A Kim S 2024723 Ornaments for efficient allele-specific expression estimation with bias correction.Am J Hum Genet 11180002-92971770178110.1016/j.ajhg.2024.06.01439047729 PMC 11339617 · doi ↗ · pubmed ↗
- 2Anders S Pyl PT Huber W 2014925 HT Seq--a Python framework to work with high-throughput sequencing data.Bioinformatics 3121367-480316616910.1093/bioinformatics/btu 63825260700 PMC 4287950 · doi ↗ · pubmed ↗
- 3Andersen EC Gerke JP Shapiro JA Crissman JR Ghosh R Bloom JS Félix MA Kruglyak L 2012129 Chromosome-scale selective sweeps shape Caenorhabditis elegans genomic diversity.Nat Genet 4431061-403628529010.1038/ng.105022286215 PMC 3365839 · doi ↗ · pubmed ↗
- 4Cutter AD Jovelin R Dey A 2013318 Molecular hyperdiversity and evolution in very large populations.Mol Ecol 2280962-10832074209510.1111/mec.1228123506466 PMC 4065115 · doi ↗ · pubmed ↗
- 5Dobin A Davis CA Schlesinger F Drenkow J Zaleski C Jha S Batut P Chaisson M Gingeras TR 20121025 STAR: ultrafast universal RNA-seq aligner.Bioinformatics 2911367-4803152110.1093/bioinformatics/bts 63523104886 PMC 3530905 · doi ↗ · pubmed ↗
- 6The ENCODE Project Consortium 201291 An integrated encyclopedia of DNA elements in the human genome Nature 48974140028-0836577410.1038/nature 1124722955616 PMC 3439153 · doi ↗ · pubmed ↗
- 7Fan J Hu J Xue C Zhang H Susztak K Reilly MP Xiao R Li M 2020511 ASEP: Gene-based detection of allele-specific expression across individuals in a population by RNA sequencing.P Lo S Genet 1651553-7390 e 1008786 e 100878610.1371/journal.pgen.100878632392242 PMC 7241832 · doi ↗ · pubmed ↗
- 8Frazee AC Jaffe AE Langmead B Leek JT 2015428 Polyester: simulating RNA-seq datasets with differential transcript expression.Bioinformatics 31171367-48032778278410.1093/bioinformatics/btv 27225926345 PMC 4635655 · doi ↗ · pubmed ↗