The genome sequence of wild privet, Ligustrum vulgare L
Zoë A. Goodwin, David Bell, Vladimir Krivtsov, Michelle L. Hart, Marco Pessoa-Filho, Irene Julca, Abhisek Chakraborty

TL;DR
This paper presents the genome sequence of wild privet, including its chromosomal structure and gene annotations.
Contribution
The study provides a high-quality genome assembly and gene annotations for Ligustrum vulgare.
Findings
The genome assembly is 1,384.00 megabases long and scaffolded into 23 chromosomal pseudomolecules.
The mitochondrial genome consists of two circularised molecules totaling 962.72 kilobases.
Gene annotation identified 31,482 protein-coding genes.
Abstract
We present a genome assembly from a specimen of Ligustrum vulgare (wild privet; Streptophyta; Magnoliopsida; Lamiales; Oleaceae). The genome sequence has a total length of 1,384.00 megabases. Most of the assembly is scaffolded into 23 chromosomal pseudomolecules. The multipartite mitochondrial genome consists of two circularised molecules with lengths of 844.62 and 118.10 kilobases, and the plastid genome assembly is 161.82 kilobases long. Gene annotation of this assembly on Ensembl identified 31,482 protein-coding genes.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1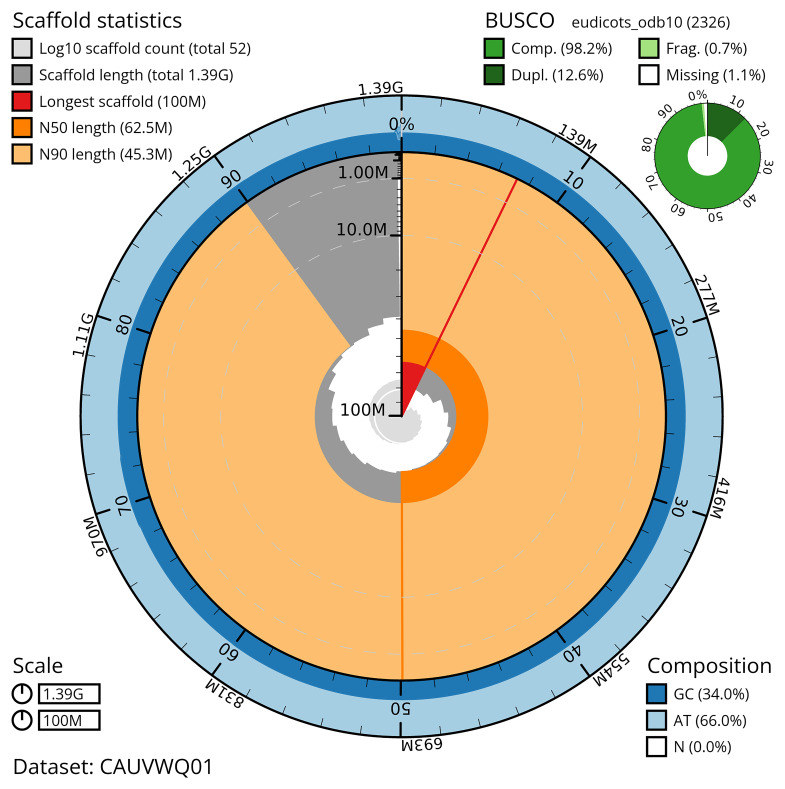 Figure 2
Figure 2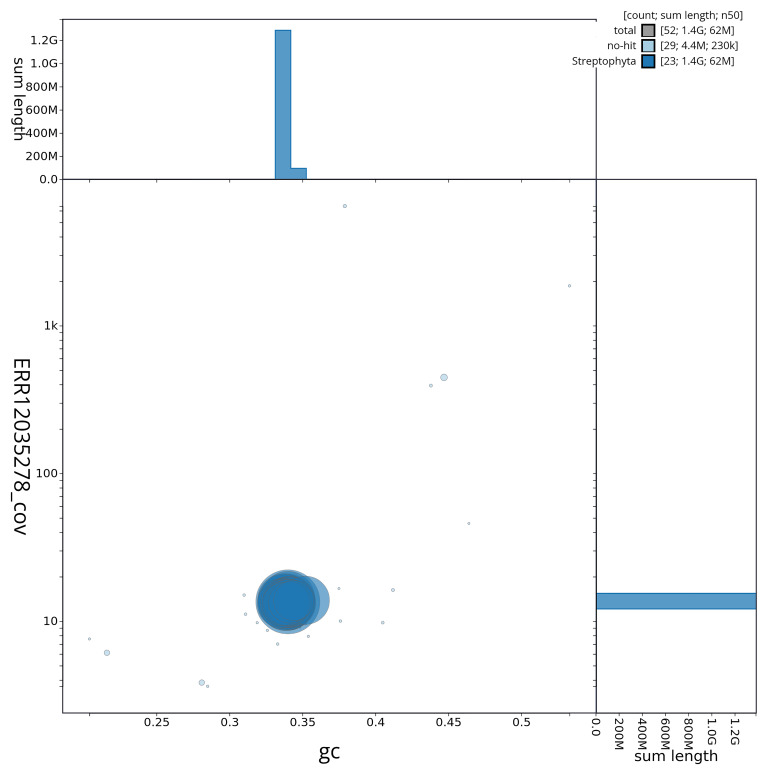 Figure 3
Figure 3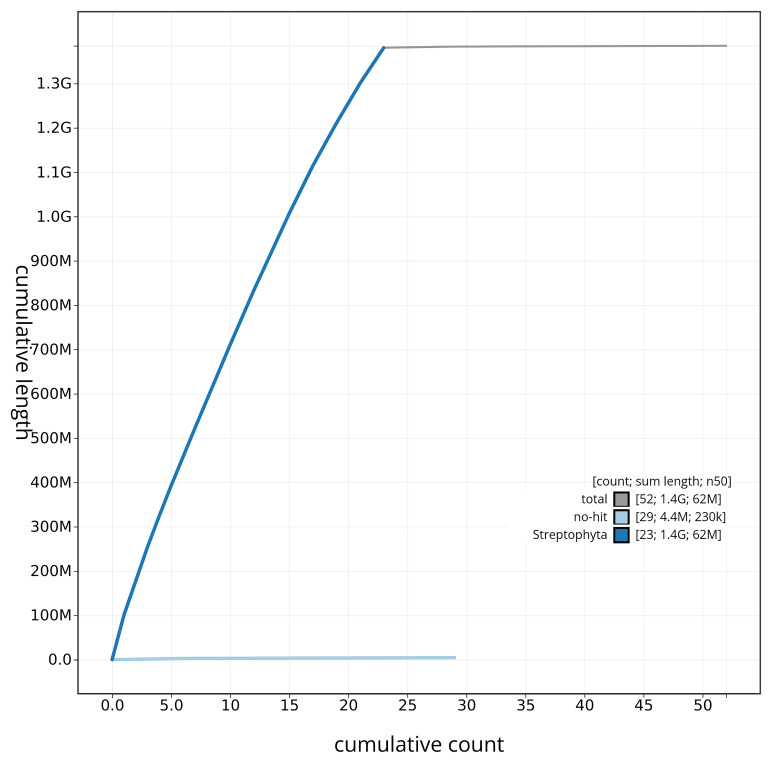 Figure 4
Figure 4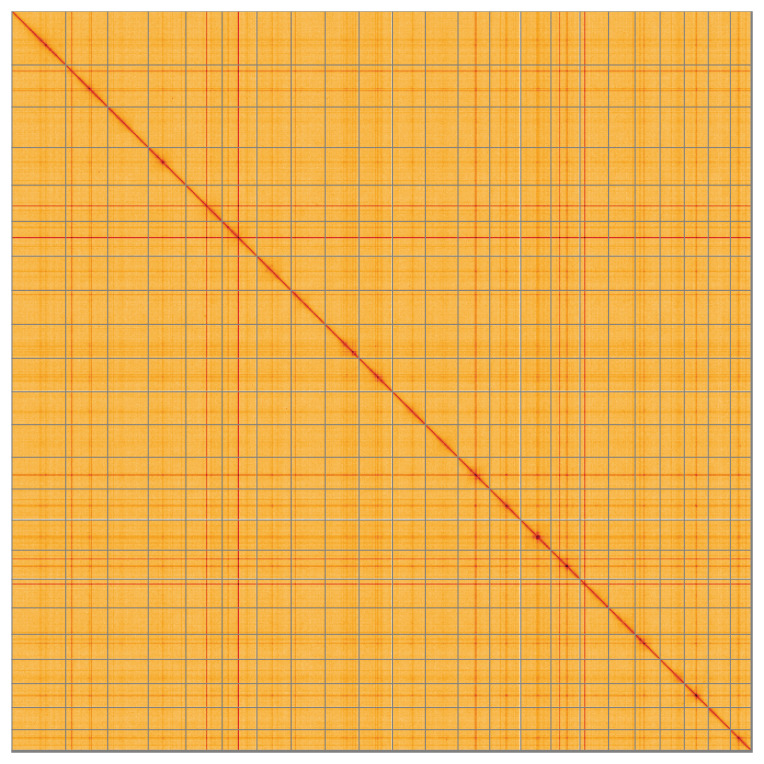 Figure 5
Figure 5| Project information | |||
|---|---|---|---|
|
| Ligustrum vulgare (common privet) | ||
|
| PRJEB65698 | ||
|
|
| ||
|
| SAMEA7535976 | ||
|
| 13597 | ||
| Specimen information | |||
|
|
|
|
|
|
| daLigVulg1 | SAMEA8596958 | leaf |
|
| daLigVulg1 | SAMEA111431662 | leaf |
|
| daLigVulg1 | SAMEA111431662 | leaf |
| Sequencing information | |||
|
|
|
|
|
|
| ERR12035282 | 1.17e+09 | 176.58 |
|
| ERR12015761 | 1.78e+06 | 26.54 |
|
| ERR12015762 | 1.61e+06 | 24.23 |
|
| ERR12015763 | 4.49e+05 | 6.18 |
|
| ERR12035283 | 6.83e+07 | 10.31 |
| Genome assembly | ||
|---|---|---|
| Assembly name | daLigVulg1.1 | |
| Assembly accession | GCA_963555705.1 | |
|
|
| |
| Span (Mb) | 1,384.00 | |
| Number of contigs | 233 | |
| Contig N50 length (Mb) | 11.4 | |
| Number of scaffolds | 49 | |
| Scaffold N50 length (Mb) | 62.5 | |
| Longest scaffold (Mb) | 100.42 | |
| Assembly metrics
|
| |
| Consensus quality (QV) | 62.2 |
|
|
| Primary: 87.81%; alternate: 84.33%; combined: 97.76% |
|
| BUSCO
| C:98.2%[S:85.7%,D:12.6%],F:0.7%,M:1.1%,n:2,326 |
|
| Percentage of assembly mapped to chromosomes | 99.68% |
|
| Organelles | Mitochondrial genome: 844.62 and 118.10 kb; plastid genome: 161.82 kb |
|
| Genome annotation at Ensembl | ||
| Number of protein-coding genes | 31,482 | |
| Number of non-coding genes | 18,273 | |
| Number of gene transcripts | 62,120 | |
| INSDC accession | Name | Length (Mb) | GC% |
|---|---|---|---|
| 1 | 100.42 | 34.0 | |
| 2 | 78.49 | 34.0 | |
| 3 | 75.69 | 34.0 | |
| 4 | 70.29 | 34.0 | |
| 5 | 67.64 | 34.0 | |
| 6 | 65.07 | 34.0 | |
| 7 | 63.98 | 33.5 | |
| 8 | 63.52 | 34.0 | |
| 9 | 63.26 | 34.0 | |
| 10 | 62.49 | 34.0 | |
| 11 | 61.41 | 34.0 | |
| 12 | 60.97 | 34.0 | |
| 13 | 59.27 | 33.5 | |
| 14 | 58.05 | 34.0 | |
| 15 | 56.32 | 35.0 | |
| 16 | 54.17 | 34.0 | |
| 17 | 53.49 | 34.0 | |
| 18 | 49.47 | 34.0 | |
| 19 | 46.75 | 34.0 | |
| 20 | 45.3 | 34.0 | |
| 21 | 44.63 | 33.5 | |
| 22 | 41.39 | 34.0 | |
| 23 | 38.65 | 34.5 | |
| Pltd | 0.16 | 38.0 | |
| MT1 | 0.84 | 44.5 | |
| MT2 | 0.12 | 44.0 |
| Software tool | Version | Source |
|---|---|---|
| BEDTools | 2.30.0 |
|
| Blast | 2.14.0 |
|
| BlobToolKit | 4.3.7 |
|
| BUSCO | 5.4.3 and 5.5.0 |
|
| bwa-mem2 | 2.2.1 |
|
| Cooler | 0.8.11 |
|
| DIAMOND | 2.1.8 |
|
| fasta_windows | 0.2.4 |
|
| FastK | 427104ea91c78c3b8b8b49f1a7d6bbeaa869ba1c |
|
| Gfastats | 1.3.6 |
|
| GoaT CLI | 0.2.5 |
|
| Hifiasm | 0.16.1-r375 |
|
| HiGlass | 44086069ee7d4d3f6f3f0012569789ec138f42b84aa44357826c0b6753eb28de |
|
| Merqury.FK | d00d98157618f4e8d1a9190026b19b471055b22e |
|
| MitoHiFi | 2 |
|
| MultiQC | 1.14, 1.17, and 1.18 |
|
| NCBI Datasets | 15.12.0 |
|
| Nextflow | 23.04.0-5857 |
|
| PretextView | 0.2.5 |
|
| OATK | 0.9 |
|
| purge_dups | 1.2.3 |
|
| samtools | 1.16.1, 1.17, and 1.18 |
|
| Seqtk | 1.3 |
|
| Singularity | 3.9.0 |
|
| YaHS | 1.1a.2 |
|
- —Wellcome Trust
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Ecology and Taxonomy Studies · Plant Taxonomy and Phylogenetics · Plant Parasitism and Resistance
Species taxonomy
Eukaryota; Viridiplantae; Streptophyta; Streptophytina; Embryophyta; Tracheophyta; Euphyllophyta; Spermatophyta; Magnoliopsida; Mesangiospermae; eudicotyledons; Gunneridae; Pentapetalae; asterids; lamiids; Lamiales; Oleaceae; Oleeae; Ligustrum; Ligustrum vulgare L. (NCBI:txid13597).
Background
The wild privet, Ligustrum vulgare L. , is a deciduous to semi-evergreen shrub native to Britain and the Channel Islands, and a neophyte in Ireland ( Stroh et al., 2023). It has a largely European distribution, occurring from Scandinavia to North Africa, and east to Iran ( WFO, 2025).
It is commonly found in hedgerows, woodland, and scrub, preferring well-drained calcareous soils ( Stroh et al., 2023). With insect-pollinated, fragrant, small white flowers from June to August, wild privet is an important food source for many insects. These include the enormous caterpillars of the privet hawk moth Sphinx ligustri ( Waring et al., 2017). The black berries are eaten and dispersed by birds ( Snow & Snow, 1988), though all parts of the plant are toxic to humans ( Cooper & Johnson, 1998). Various parts of Ligustrum vulgare are demonstrated to have medicinal potential, including strong antiproliferative properties for the treatment of cancer ( Litewski et al., 2024). Like the more reliably evergreen privet, Ligustrum ovalifolium Hassk., wild privet is commonly used in garden hedging.
Here we present a chromosomally complete genome sequence for Ligustrum vulgare, based on a plant growing at the Royal Botanic Garden Edinburgh (Inverleith), Midlothian, Scotland, UK that was originally collected from Epsom Downs, Surrey. The assembled genome presented here is consistent with published chromosome counts of 2 n = 46 ( BSBI, 2025).
Genome sequence report
The genome size (1C-value) of the Ligustrum vulgare specimen ( Figure 1) was estimated to be 1.71 pg, equivalent to 1,670 Mb, by flow cytometry. The genome was sequenced using Pacific Biosciences single-molecule HiFi long reads, generating a total of 6.18 Gb (gigabases) from 0.45 million reads, providing approximately 34-fold coverage. Primary assembly contigs were scaffolded with chromosome conformation Hi-C data, which produced 176.58 Gb from 1,169.42 million reads. Specimen and sequencing details are provided in Table 1.
Photograph of the Ligustrum vulgare (daLigVulg1) specimen used for genome sequencing.
Table 1.: Specimen and sequencing data for Ligustrum vulgare.
Manual assembly curation corrected 48 missing joins or mis-joins, reducing the scaffold number by 32.47%, and increasing the scaffold N50 by 1.25%. The final assembly has a total length of 1,384.00 Mb in 49 sequence scaffolds, with 181 gaps and a scaffold N50 of 62.5 Mb ( Table 2) The snail plot in Figure 2 provides a summary of the assembly statistics, while the distribution of assembly scaffolds on GC proportion and coverage is shown in Figure 3. The cumulative assembly plot in Figure 4 shows curves for subsets of scaffolds assigned to different phyla. Most (99.68%) of the assembly sequence was assigned to 23 chromosomal-level scaffolds. Chromosome-scale scaffolds confirmed by the Hi-C data are named in order of size ( Figure 5; Table 3). There appears to be a heterozygous translocation between chromosomes 20 and 21. While not fully phased, the assembly deposited is of one haplotype. Contigs corresponding to an alternate haplotype have also been deposited. The mitochondrial and plastid genomes were also assembled and can be found as contigs within the multifasta file of the genome submission.
Table 2.: Genome assembly data for Ligustrum vulgare, daLigVulg1.1.
Genome assembly of Ligustrum vulgare, daLigVulg1.1: metrics.The BlobToolKit snail plot shows N50 metrics and BUSCO gene completeness. The main plot is divided into 1,000 size-ordered bins around the circumference with each bin representing 0.1% of the 1,385,159,111 bp assembly. The distribution of scaffold lengths is shown in dark grey with the plot radius scaled to the longest scaffold present in the assembly (100,421,371 bp, shown in red). Orange and pale-orange arcs show the N50 and N90 scaffold lengths (62,494,563 and 45,303,176 bp), respectively. The pale grey spiral shows the cumulative scaffold count on a log scale with white scale lines showing successive orders of magnitude. The blue and pale-blue area around the outside of the plot shows the distribution of GC, AT and N percentages in the same bins as the inner plot. A summary of complete, fragmented, duplicated and missing BUSCO genes in the eudicots_odb10 set is shown in the top right. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/CAUVWQ01/dataset/CAUVWQ01/snail.
Genome assembly of Ligustrum vulgare, daLigVulg1.1: Blob plot of base coverage in ERR12035278 against GC proportion for sequences in the assembly.Sequences are coloured by phylum. Circles are sized in proportion to sequence length. Histograms show the distribution of sequence length sum along each axis. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/CAUVWQ01/dataset/CAUVWQ01/blob.
Genome assembly of Ligustrum vulgare daLigVulg1.1: BlobToolKit cumulative sequence plot.The grey line shows cumulative length for all sequences. Coloured lines show cumulative lengths of sequences assigned to each phylum using the buscogenes taxrule. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/CAUVWQ01/dataset/CAUVWQ01/cumulative.
Genome assembly of Ligustrum vulgare, daLigVulg1.1: Hi-C contact map of the daLigVulg1.1 assembly, visualised using HiGlass. Chromosomes are shown in order of size from left to right and top to bottom. An interactive version of this figure may be viewed at https://genome-note-higlass.tol.sanger.ac.uk/l/?d=LiFrhIQ8RmSFRX2IFLesUw.
Table 3.: Chromosomal pseudomolecules in the genome assembly of Ligustrum vulgare, daLigVulg1.
The estimated Quality Value (QV) of the final assembly is 62.2 with k-mer completeness of 97.76% (combined primary and alternate). The primary assembly has a BUSCO v5.4.3 completeness of 98.2% (single = 85.7%, duplicated = 12.6%), using the eudicots_odb10 reference set ( n = 2,326).
Genome annotation report
The Ligustrum vulgare genome assembly (GCA_963555705.1) was annotated at the European Bioinformatics Institute (EBI) on Ensembl Rapid Release. The resulting annotation includes 62,120 transcribed mRNAs from 31,482 protein-coding and 18,273 non-coding genes ( Table 2; https://rapid.ensembl.org/Ligustrum_vulgare_GCA_963555705.1/Info/Index). The average transcript length is 3,206.44. There are 1.25 coding transcripts per gene and 4.31 exons per transcript.
Methods
Sample acquisition, DNA barcoding and genome size estimation
Leaves from a Ligustrum vulgare shrub (specimen ID EDTOL00064, ToLID daLigVulg1) were collected from cultivated material at the Royal Botanic Garden Edinburgh (Inverleith), Midlothian, Scotland, UK (latitude 55.97, longitude –3.21), from a plant that was originally wild-collected by William Tait in 1984, from Epsom Downs, Surrey, England. Samples were collected on 2020-08-11 and on 2022-08-08. The samples were taken by Zoë Goodwin, David Bell and Vladimir Krivtsov (Royal Botanic Garden Edinburgh) and preserved by snap freezing in liquid nitrogen. The herbarium voucher from the sequenced plant is kept at the Royal Botanic Garden Edinburgh (E) https://data.rbge.org.uk/herb/E01358645.
The initial species identification was verified by an additional DNA barcoding process according to the framework developed by Twyford et al. (2024). Part of the plant specimen was preserved in silica gel desiccant. A DNA extraction from the dried plant was amplified by PCR for standard barcode markers, with the amplicons sequenced and compared to public sequence databases including GenBank and the Barcode of Life Database (BOLD). The barcode sequences for this specimen are openly available on BOLD ( Ratnasingham & Hebert, 2007). Following whole genome sequence generation, DNA barcodes were also used alongside the initial barcoding data for sample tracking through the genome production pipeline at the Wellcome Sanger Institute ( Twyford et al., 2024). The standard operating procedures for the Darwin Tree of Life barcoding have been deposited on protocols.io ( Beasley et al., 2023).
The genome size was estimated by flow cytometry using the fluorochrome propidium iodide and following the ‘one-step’ method as outlined in Pellicer et al. (2021). For this species, the General Purpose Buffer (GPB) supplemented with 3% PVP and 0.08% (v/v) beta-mercaptoethanol was used for isolation of nuclei ( Loureiro et al., 2007), and the internal calibration standard was Petroselinum crispum ‘Champion Moss Curled’ with an assumed 1C-value of 2,200 Mb ( Obermayer et al., 2002).
Nucleic acid extraction
The workflow for high molecular weight (HMW) DNA extraction at the Wellcome Sanger Institute (WSI) Tree of Life Core Laboratory includes a sequence of core procedures: sample preparation; sample homogenisation, DNA extraction, fragmentation, and clean-up. In sample preparation, the daLigVulg1 sample was weighed and dissected on dry ice ( Jay et al., 2023). For sample homogenisation, leaf tissue was cryogenically disrupted using the Covaris cryoPREP ^®^ Automated Dry Pulverizer ( Narváez-Gómez et al., 2023). HMW DNA was extracted using the Automated Plant MagAttract v2 protocol ( Todorovic et al., 2023). HMW DNA was sheared into an average fragment size of 12–20 kb in a Megaruptor 3 system ( Bates et al., 2023). Sheared DNA was purified by solid-phase reversible immobilisation ( Strickland et al., 2023): in brief, the method employs a 1.8X ratio of AMPure PB beads to sample to eliminate shorter fragments and concentrate the DNA. The concentration of the sheared and purified DNA was assessed using a Nanodrop spectrophotometer and Qubit Fluorometer and Qubit dsDNA High Sensitivity Assay kit. Fragment size distribution was evaluated by running the sample on the FemtoPulse system.
RNA was extracted from leaf tissue of daLigVulg1 in the Tree of Life Laboratory at the WSI using the RNA Extraction: Automated MagMax™ mirVana protocol ( do Amaral et al., 2023). The RNA concentration was assessed using a Nanodrop spectrophotometer and a Qubit Fluorometer using the Qubit RNA Broad-Range Assay kit. Analysis of the integrity of the RNA was done using the Agilent RNA 6000 Pico Kit and Eukaryotic Total RNA assay.
Sequencing
Pacific Biosciences HiFi circular consensus DNA sequencing libraries were constructed according to the manufacturers’ instructions. Poly(A) RNA-Seq libraries were constructed using the NEB Ultra II RNA Library Prep kit. DNA and RNA sequencing was performed by the Scientific Operations core at the WSI on Pacific Biosciences Sequel IIe (HiFi) and Illumina NovaSeq 6000 (RNA-Seq) instruments. Hi-C data were also generated from leaf tissue of daLigVulg1 using the Arima-HiC v2 kit. The Hi-C sequencing was performed using paired-end sequencing with a read length of 150 bp on the Illumina NovaSeq 6000 instrument.
Genome assembly, curation and evaluation
** Assembly **
The original assembly of HiFi reads was performed using Hifiasm ( Cheng et al., 2021) with the --primary option. Haplotypic duplications were identified and removed with purge_dups ( Guan et al., 2020). Hi-C reads were further mapped with bwa-mem2 ( Vasimuddin et al., 2019) to the primary contigs, which were further scaffolded using the provided Hi-C data ( Rao et al., 2014) in YaHS ( Zhou et al., 2023) using the --break option. Scaffolded assemblies were evaluated using Gfastats ( Formenti et al., 2022), BUSCO ( Manni et al., 2021) and MERQURY.FK ( Rhie et al., 2020). The organelle genomes were assembled using OATK ( Zhou, 2023).
** Curation **
The assembly was decontaminated using the Assembly Screen for Cobionts and Contaminants (ASCC) pipeline (article in preparation). Manual curation was primarily conducted using PretextView ( Harry, 2022), with additional insights provided by JBrowse2 ( Diesh et al., 2023) and HiGlass ( Kerpedjiev et al., 2018). Scaffolds were visually inspected and corrected as described by Howe et al. (2021). Any identified contamination, missed joins, and mis-joins were corrected, and duplicate sequences were tagged and removed. The process is documented at https://gitlab.com/wtsi-grit/rapid-curation (article in preparation).
** Evaluation of assembly quality **
The Merqury.FK tool ( Rhie et al., 2020), run in a Singularity container ( Kurtzer et al., 2017), was used to evaluate k-mer completeness and assembly quality for the primary and alternate haplotypes using the k-mer databases ( k = 31) computed prior to genome assembly. The analysis outputs included assembly QV scores and completeness statistics.
A Hi-C contact map was produced for the final version of the assembly. The Hi-C reads were aligned using bwa-mem2 ( Vasimuddin et al., 2019) and the alignment files were combined using SAMtools ( Danecek et al., 2021). The Hi-C alignments were converted into a contact map using BEDTools ( Quinlan & Hall, 2010) and the Cooler tool suite ( Abdennur & Mirny, 2020). The contact map was visualised in HiGlass ( Kerpedjiev et al., 2018).
Table 4 contains a list of relevant software tool versions and sources.
Genome annotation
The Ensembl Genebuild annotation system ( Aken et al., 2016) was used to generate annotation for the Ligustrum vulgare assembly (GCA_963555705.1) in Ensembl Rapid Release at the EBI. Annotation was created primarily through alignment of transcriptomic data to the genome, with gap filling via protein-to-genome alignments of a select set of proteins from UniProt ( UniProt Consortium, 2019).
Wellcome Sanger Institute – Legal and Governance
The materials that have contributed to this genome note have been supplied by a Darwin Tree of Life Partner. The submission of materials by a Darwin Tree of Life Partner is subject to the ‘Darwin Tree of Life Project Sampling Code of Practice’, which can be found in full on the Darwin Tree of Life website here. By agreeing with and signing up to the Sampling Code of Practice, the Darwin Tree of Life Partner agrees they will meet the legal and ethical requirements and standards set out within this document in respect of all samples acquired for, and supplied to, the Darwin Tree of Life Project.
Further, the Wellcome Sanger Institute employs a process whereby due diligence is carried out proportionate to the nature of the materials themselves, and the circumstances under which they have been/are to be collected and provided for use. The purpose of this is to address and mitigate any potential legal and/or ethical implications of receipt and use of the materials as part of the research project, and to ensure that in doing so we align with best practice wherever possible. The overarching areas of consideration are:
• Ethical review of provenance and sourcing of the material
• Legality of collection, transfer and use (national and international)
Each transfer of samples is further undertaken according to a Research Collaboration Agreement or Material Transfer Agreement entered into by the Darwin Tree of Life Partner, Genome Research Limited (operating as the Wellcome Sanger Institute), and in some circumstances other Darwin Tree of Life collaborators.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abdennur N Mirny LA : Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 2020;36(1):311–316. 10.1093/bioinformatics/btz 540 31290943 PMC 8205516 · doi ↗ · pubmed ↗
- 2Aken BL Ayling S Barrell D : The Ensembl gene annotation system. Database (Oxford). 2016;2016: baw 093. 10.1093/database/baw 093 27337980 PMC 4919035 · doi ↗ · pubmed ↗
- 3Bates A Clayton-Lucey I Howard C : Sanger Tree of Life HMW DNA fragmentation: diagenode Megaruptor ®3 for LI Pac Bio. protocols.io. 2023. 10.17504/protocols.io.81wgbxzq 3lpk/v 1 · doi ↗
- 4Beasley J Uhl R Forrest LL : DNA barcoding SO Ps for the Darwin Tree of Life project. protocols.io. 2023; [Accessed 25 June 2024]. 10.17504/protocols.io.261ged 91jv 47/v 1 · doi ↗
- 5BSBI: BSBI cytology database.2025; [Accessed 19 March 2025]. Reference Source
- 6Cheng H Concepcion GT Feng X : Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 2021;18(2):170–175. 10.1038/s 41592-020-01056-5 33526886 PMC 7961889 · doi ↗ · pubmed ↗
- 7Cooper MR Johnson AW : Poisonous plants and fungi in Britain: animal and human poisoning. 2nd edition. London, UK: The Stationery Office,1998. Reference Source
- 8Danecek P Bonfield JK Liddle J : Twelve years of SA Mtools and BC Ftools. Giga Science. 2021;10(2): giab 008. 10.1093/gigascience/giab 008 33590861 PMC 7931819 · doi ↗ · pubmed ↗