A descriptive analysis of sporadic Creutzfeldt-Jakob cases in Vietnam: 31 patients from four tertiary care centers
Khoi Hong Vo, Nga Thi Bui, Dung Thi Hoang, Thanh Vinh Nguyen, Binh Thanh Nguyen, Tai Ngọc Tran, Anh Hai Nguyen, My Thi Le, Ha Thi Do, Thuan Duc Nguyen, Hai Van Ta, Toan Van Phan, Phuc Duy Phan, Thang Xuan Pham, Frank Xing, Daniel Truong

TL;DR
This study provides the first systematic analysis of 31 sporadic Creutzfeldt-Jakob disease cases in Vietnam, describing clinical features and diagnostic challenges.
Contribution
The first systematic analysis of sporadic Creutzfeldt-Jakob disease in Vietnam, highlighting diagnostic patterns and clinical features.
Findings
Most patients presented with rapidly progressive dementia and myoclonus, with an average survival of 7.6 months.
Neuroimaging and EEG findings were consistent with global sCJD patterns, including FLAIR/DWI abnormalities and periodic sharp wave complexes.
Only 12 patients were tested for 14–3-3 protein in CSF, and all tested positive.
Abstract
Accurate diagnosis of sCJD remains challenging in developing countries such as Vietnam, as clinical and research efforts focus on treatable diseases. Several cases of sCJD have been diagnosed in Vietnam but data from formal analyses are lacking. This is the first systematic analysis of patients with sCJD in Vietnam. This was a systematic retrospective review of medical records from patients with probable sCJD (N = 31) seen between April 2021 to April 2024 at four tertiary care centers. Clinical, laboratory, neuroimaging, and EEG findings were included in the analysis. Data from 16 men and 15 women with sCJD were analyzed. The average age of onset was 63.4 years (range 50–83 years). Twenty-one of the patients had died after a mean survival of 7.6 months (range 3–20 months). All patients initially presented with rapidly progressive dementia. Other associated symptoms included myoclonus…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
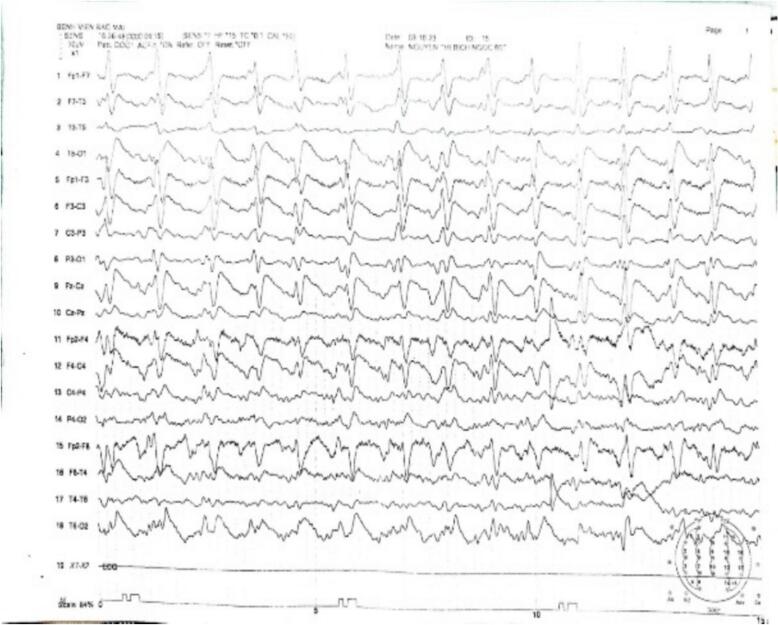 Figure 1
Figure 1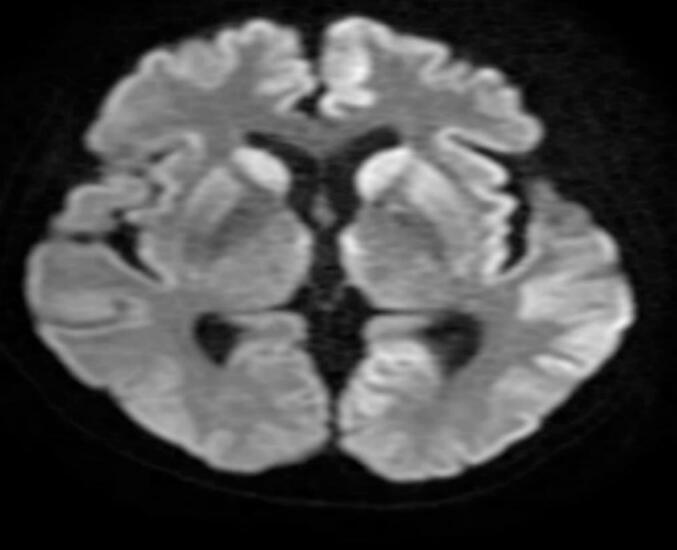 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPrion Diseases and Protein Misfolding
Introduction
1
Creutzfeldt-Jakob disease (CJD) is a fatal, neurodegenerative prion disease with an annual global incidence of approximately 1–2 million people [1,2]. CJD has a variable clinical presentation at onset and rapid progression, with a mean survival of 4 to 6 months [1,2]. Epidemiologic data suggest that the incidence of CJD may be on the rise, but it remains unknown whether this increase reflects a true rise in rates of disease or is related to factors such as aging of the population, better recognition, better diagnostic tools, or other factors [1,2]. Historically, diagnoses of CJD could only be made post-mortem, but more recent tools and methods have led to earlier diagnosis.
Complicating the early recognition and diagnosis of CJD is the fact that at least six distinct molecular subtypes have been identified, which vary in time of onset, survival, and neuropathological findings [1]. Sporadic CJD (sCJD) is the most common type of CJD, accounting for up to 85 % of cases [2]. Early diagnosis of sCJD can also be challenging due to its nonspecific clinical presentation and overlap with symptoms of other neurological and psychiatric conditions [3]. As a result, diagnosis often occurs when the disease is already advanced and the window for potential experimental therapies has closed [1].
Although sCJD has been extensively studied in many countries, in part with the assistance of surveillance infrastructure, its epidemiology is still poorly understood outside of North America, Europe, and some countries in Asia [4]. In Asia specifically, CJD is likely underreported in some areas due to regional disparities in surveillance and reporting [5]. Furthermore, data suggest that recognition and diagnosis are challenging and difficult to study in low- and lower-middle-income countries [4].
The goal of this retrospective descriptive analysis was to characterize patients with sCJD in Vietnam, which, to date, have largely been described in case studies. This research represents the first systematic report of the clinical, laboratory, neuroimaging, and electroencephalographic characteristics of patients with sCJD in Vietnam, utilizing data collected from four tertiary care hospitals. Better understanding of the initial and later presentation of these patients may help to improve diagnosis and recognition in this country.
Methods
2
Study design and patient population
2.1
This retrospective analysis was conducted on data collected from medical records of patients with sCJD treated at four tertiary care hospitals from April 2021 to April 2024: Bach Mai Hospital (Hanoi), Military Hospital 103 (Ho Chi Minh City), Hanoi Medical University Hospital, and University Medical Center Ho Chi Minh City. This study received ethical approval from the Biomedical Research Ethics Committee of Bach Mai Hospital.
Researchers at each medical center collected data from patients seen at their hospital using a customized research template. Electronic medical records were searched for diagnoses containing the key term “sporadic Creutzfeldt-Jakob Disease”. We found 36 patients with this diagnosis. However, only 31 patients met the United States Centers for Disease Control and Prevention 2010 diagnostic criteria for probable sCJD; a definitive diagnosis requires brain biopsy [6,7]. Criteria for probable sCJD include clinical manifestations of progressive dementia and at least two of the following four signs: myoclonus, visual or cerebellar disease, pyramidal or extrapyramidal symptoms, and akinetic mutism, as well as characteristic changes in electroencephalogram (EEG) and magnetic resonance imaging (MRI), or laboratory data, such as positive 14–3-3 protein in cerebrospinal fluid (CSF). Moreover, routine investigations must not suggest an alternative diagnosis. The remaining 5 cases identified by searching had missing information that precluded a diagnosis (MRI, EEG, lumbar puncture, or tests to rule out metabolic diseases). Of the final 31 cases, 18 were from Bach Mai Hospital, 8 were from the University of Medicine and Pharmacy Ho Chi Minh City, 2 were from 103 Hospital, and 3 were from Hanoi Medical University Hospital. A trained neurologist reviewed patient records and pertinent information extracted for the analysis.
Data collection and analysis
2.2
Baseline demographic, clinical, laboratory, MRI, and EEG data were abstracted from patient medical records and recorded on a custom research form. Specific clinical symptoms and signs associated with sCJD were collected from both subjective and objective portions of the medical record. Symptom onset was recorded as of the initial hospital visit. Symptoms after disease progression were recorded from the time of hospitalization to discharge. Results of CSF testing for protein 14–3-3 were available for 12 patients and were also recorded and included in the analysis. Survival time was defined as the time from disease onset until death. Follow-up information regarding deceased patients was obtained through phone calls with the patients' families.
Information was collected on cerebellar symptoms, defined as evidence of dysmetria on the finger-to-nose or heel-knee-shin test and nystagmus on eye exam; pyramidal signs, including hyperreflexia (positive Babinski sign or documented muscle spasticity); extrapyramidal signs including rigidity, tremor, bradykinesia, restless legs syndrome or dystonia; epilepsy based on clinical diagnosis with or without epileptiform spike-wave activity on EEG; neuropsychiatric symptoms of depression, fretfulness, anxiety, autism, apathy, insomnia, obsession, excitement, abnormal emotion, character change, abnormal behavior or memory disorder; and sleep disorders diagnosed clinically based on the presence of insomnia, lack of sleep, or hypersomnia. Polysomnography data was not available for any patients included in the analysis.
Data from two commonly used and well-validated assessments, the Mini Mental Status Exam (MMSE; [8]) and the Medical Research Council scale (MRC; [9]) were also collected and analyzed for this study. The MMSE is a 30-question clinician administered assessment of five areas of cognitive function: orientation, registration, attention, and calculation, recall, and language. Each question is rated as 0 or 1 for a maximum possible score of 30, with higher scores reflecting better cognitive function. A score of < 24 was considered indicative of dementia. Rapidly progressive dementia (RPD) was defined as onset of dementia within 1–=–2 years of disease onset [10]. The MRC is a milestone-based functional scale with both language and non-language components [9] used to evaluate dynamic aphasia characteristic of prion disease. The verbal component is a score of 0 (mute) to 4 (normal conversation), intended to identify gross onset and progression of language disturbance. An MRC score of < 4 was used to define the presence of a language disorder.
Objective findings included brain MRI, EEG, and lumbar puncture. All MRIs were performed using a 1.5-T superconducting magnet (GE Healthcare, Pittsburgh, PA, USA) and included T1, T2, fluid-attenuated inversion recovery (FLAIR), and diffusion-weighted imaging (DWI) sequences. All EEGs were performed using Nihon Kohden EEG-1200 K.
Descriptive data are reported. No statistical analysis was conducted.
Results
3
Demographic and clinical characteristics
3.1
Sixteen men and 15 women were included in this descriptive analysis. The mean age at disease onset was 63.4 years (range 50–83 years). Mean time to diagnosis of sCJD was 2.7 months (range 1–14 months) and mean time from diagnosis to death was 4.9 months (range 0.5–15.5 months). Twenty-one patients had been misdiagnosed at the primary hospital before transferring to the university hospital. Misdiagnoses included Parkinson's syndrome (n = 7), Alzheimer's disease (n = 6), psychosis (n = 4), encephalitis (n = 3), and epilepsy (n = 1).
Table 1 shows the most common symptoms at disease onset and following disease progression. The most common symptom at disease onset was RPD (84 %). Language disorders, extrapyramidal signs, visual disturbances, and neuropsychiatric disorders were reported in 29 % to 35 % of patients.Table 1. Clinical symptoms at onset and after disease progression.CharacteristicNumber of patients (N = 31)Symptoms at onsetaDementia (MMSE < 24)26Language disorders (MRC > 4)26Extrapyramidal symptoms11Neuropsychiatric symptoms9Visual disturbance8Vertigo, balance disorders7Symptoms after disease progressionbDementia (MMSE < 24)31Extrapyramidal symptoms25Myoclonus24Pyramidal symptoms18Akinetic mutism17Visual disturbance14Neuropsychiatric symptomsc12Cerebellar symptoms10Sleep disorders9Epilepsyd8MMSE, Mini Mental State Exam; MRC, Medical Research Council scale.aOnset symptoms are symptoms recorded as of the initial hospital day visit.bSymptoms following disease progression were recorded from hospitalization until discharge.cNeuropsychiatric disorders included hallucinations, delusions, and mood disorders.dTwo patients presented with both epilepsy and sleep disorders.
Following disease progression, the most frequent clinical manifestations were continuing RPD (100 %), myoclonus (77 %), and extrapyramidal symptoms including parkinsonism and dystonia (81 %). Pyramidal symptoms, visual disturbance, and akinetic mutism were also common, as shown in Table 1. Other neurologic symptoms included cerebellar symptoms, epilepsy, neuropsychiatric symptoms, and sleep disorders were also noted (Table 1).
MRI and EEG findings
3.2
All patients included in this analysis had undergone MRI and EEG. Medical records review revealed that 28/31 patients (90 %) had hyperintensities on DWI and/or FLAIR sequences in the cortical and basal ganglia regions of the brain. In patients with hyperintensities, lesions were documented in the cortex in all 28 patients, the basal ganglia in 19 patients, or both (n = 19; Table 2).Table 2. Key clinical, neuroimaging, and survival findings.FindingsPatientsBrain MRI**n = 31Abnormality (any), n28Cortical abnormality, n28Basal ganglia abnormality, n19DWI hyperintensity, n26FLAIR hyperintensity, n28EEG**n = 31PSWC positive, n26Diffuse slow waves, n16Epileptiform discharges, n14CSF**n = 31Elevated WBC count (>5 cells/µl), n4Elevated total protein (>0.5 g/l), n514–3-3 protein**n = 12Positive, n12Time of symptom onset in protein-positive patients, mean (range) weeks10 (5–28)Survival**n = 21Survival time (time from onset to death), mean (range), months7.6 (3–20)Survival time ≤ 1 year, n17CSF, cerebrospinal fluid; DWI, diffusion-weighted imaging; EEG, electroencephalogram; FLAIR, fluid-attenuated inversion recovery; EEG, electroencephalogram; MRI, magnetic resonance imaging; PSWC, periodic sharp wave complexes; WBC, white blood cell.
Twenty-six patients (84 %) had evidence of periodic sharp wave complexes (PSWCs). Other abnormal EEG patterns included focal epileptiform discharges and diffuse slow waves as shown in Table 2.
In 3 patients with normal MRI findings, EEG showed PSWCs. In 5 patients with normal EEG, brain MRI revealed lesions.
Fig. 1, Fig. 2 show EEG and imaging findings, respectively, from a 61-year-old Vietnamese patient who presented with dementia, myoclonus, neuropsychiatric, pyramidal, and extrapyramidal symptoms. On hospital day 5, EEG revealed PSWCs on EEG and MRI DWI hyperintensities in the bilateral caudate, putamen and cortex.Fig. 1. Electroencephalographic findings in a patient with probable sporadic Creutzfeldt-Jakob disease.Fig. 2. Diffusion-weighted MRI (DWI) shows hyperintensities in the bilateral caudate, putamen, and cortical regions in a Vietnamese patient with probably sporadic Cruetzfeld-Jakob disease.
CSF 14–3-3 protein
3.3
Twelve patients had data from CSF 14–3-3 protein testing, all demonstrating positive results. The mean time from symptoms onset to 14–3-3 protein positivity was 10 weeks (range 5–28 weeks; Table 2).
Survival
3.4
At the time we conducted this analysis, 10 patients were still alive. Twenty-one of the 31 patients in our analysis had died prior to our data extraction. Patients had a mean survival time of 7.6 months (range 3–20 months). Among these patients, 17 cases (55 %) survived < 1 year and 4 cases ≥ 1 year (Table 2).
Discussion
4
This retrospective, descriptive analysis of clinical symptoms, supportive diagnostic tests, and patient survival time provides the first comprehensive description of patients with sCJD in Vietnam. Results show that characteristics of patients with sCJD in Vietnam align with those expected based on diagnostic criteria and in most respects are similar to those reported for China [[11], [12]] and other parts of Asia [[13], [14], [15]], Europe [13], and the United States [14]. However, the relatively small number of patients identified in our analysis suggests that neither patients nor physicians are routinely able to identify early symptoms of disease. Further supporting that notion, 5 patients diagnosed with sCJD by their local neurologists did not, in fact meet CDC criteria for probable disease [6], and 21 of 31 patients were initially misdiagnosed.
There are number of possible reasons for delayed, incorrect, or missed diagnosis of sCJD in Vietnam. First, the number of neurologists practicing in Vietnam is approximately 0.48 per 100,000 people based on 2007 data [16]. Second, patients have difficulty accessing neurologists, who are often concentrated at the central hospital. Third, because of a cultural bias in Vietnam to focus limited medical resources on curable diseases, available neurologists are likely to be inadequately trained to identify sCJD or may not be looking for it. Finally, some primary care facilities have limited or no access to ancillary testing such as MRI, EEG, or CSF testing, which are required for probable diagnosis.
Definitive diagnosis of sCJD is even less likely in Vietnam, as it requires brain biopsy and neuropathological examination. Brain biopsy carries inherent risks and potential complications and are therefore generally unacceptable to Vietnamese patients given the fact that biopsy cannot inform or improve prognosis. Brain biopsies are commonly performed in Vietnam to help diagnose and treat brain tumors with the hope of positive outcome.
This study analyzing 31 patients with probable sCJD in Vietnam revealed the need for improved education and awareness about sCJD both so early symptoms can be monitored and to provide a more accurate estimate of the epidemiology of sCJD in the country. Our patients showed both similarities and differences compared to other countries. The average survival time for sCJD patients in Vietnam was shorter compared to those in China, the United States, and Japan (Table 3). This is likely due to patients being diagnosed later in the disease course when symptoms are more severe. Our study reported higher rates of PSWCs, MRI, and CSF 14–3-3 protein abnormalities compared to studies in China, Europe, and the United States (Table 3). The availability of advanced diagnostic methods, such as RT-QuIC and brain biopsy, was limited in this study. These tests should be more widely accessible and utilized to enhance diagnostic accuracy. In the interim, additional studies of patients in Vietnam should be conducted to continue to characterize patient characteristics and to determine whether surveillance data from other Asian countries may help to inform earlier and more accurate diagnosis.Table 3. Regional comparison of epidemiology, patient demographics, diagnosis, and clinical presentation of sCJD**CountryOur StudyVietnam (n = 31)**Eastern India [15] **(n = 10)**Thailand [17] **(n = 18)**Japan [18] **(n = 674)**China[11] **(n = 104)China[12](n = 67)**Europe [13] (n = 2451)United States [14](n = 116)DemographicsGender (% male)5240444354.871.645.767.2Age of onset [mean (range) years]64.4(50–83)56.1(39–70)66.766.760 (29–82)64.42(29–88)67.2(15.6–94.9)65Survival time [mean (range) months]7.7(3–20)4.6NR15.79 (1–36)9.39 (1–60)5 (1–81)10.6Survival time ≤ 1 year (%)80.9NRNRNRNR79.485.8NRSurvival time > 1 year19.1NRNRNRNR20.614.2NRInitial misdiagnosis (%)67.7NRNRNR56.7NRNRNRClinical symptoms (% of patients)Rapid progressive dementia100100100NR10050.7NR95Myoclonus77.410077.8NRNR70.1NR63Extrapyramidal80.65055.6NR37.2NRNR65Akinetic mutism54.810044.4NR45.237.3NRNRPyramidal58.1NR55.6NR43.338.8NRNRVisual disturbance45.25027.813.631.725.3NR64Cerebellar ataxia32.24050NR51.970.1NR81Epilepsy25.8NRNRNRNRNRNRNRNeuropsychiatric38.76033.3NR7.758.2NR41Sleep disorders29NR44.4NR4.8NRNRNRKey testing features*Periodic sharp wave complexes (%)83.86066.791.9674658.435Abnormal MRI (%)90.310094NR88.284.839.164Cortex (%)90.3NR38.9NR82.475.7NR31Basal ganglia (%)61.3NR66.7NR38.275.7NR51CSF 14–3-3 protein positive (%)100NRNRNR34.16888.171Time of symptom onset to 14–3-3 protein positive (mean, weeks)10(5–28)NRNRNRNRNRNR21.4Bisophy (%)0NRNRNR9.6NRNR16Autophy (%)0NRNRNR1NRNRNRNR=not reported
CRediT authorship contribution statement
Khoi Hong Vo: Writing – original draft, Supervision, Resources, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Nga Thi Bui: Writing – original draft, Methodology, Investigation, Formal analysis, Data curation. Dung Thi Hoang: Writing – review & editing, Writing – original draft, Resources, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Thanh Vinh Nguyen: Writing – original draft, Resources, Investigation, Data curation. Binh Thanh Nguyen: Writing – original draft, Supervision, Resources, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Tai Ngọc Tran: Writing – original draft, Resources, Investigation, Data curation. Anh Hai Nguyen: Writing – original draft, Resources, Investigation, Data curation. My Thi Le: Writing – original draft, Resources, Investigation, Data curation. Ha Thi Do: Writing – original draft. Thuan Duc Nguyen: Writing – original draft, Resources, Investigation, Data curation. Hai Van Ta: Writing – original draft, Resources, Investigation. Toan Van Phan: Writing – original draft, Resources, Investigation, Data curation. Phuc Duy Phan: Writing – original draft, Resources, Investigation, Data curation. Thang Xuan Pham: Writing – original draft, Resources, Investigation, Data curation. Frank Xing: Writing – review & editing, Writing – original draft, Resources, Investigation, Data curation. Daniel Truong: Writing – review & editing, Writing – original draft, Supervision, Resources, Methodology, Investigation, Formal analysis, Data curation, Conceptualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jurcau M.C.Jurcau A.Diaconu R.G.Hogea V.O.Nunkoo V.S.A systemiatic review of sporadic Creutzfeldt-Jakob disease: pathogenesis, diagnosis, and therapeutic attempts Neurol Int 1652024103910653931135210.3390/neurolint 16050079 PMC 11417857 · doi ↗ · pubmed ↗
- 2Uttley L.Carroll C.Wong R.Hilton D.A.Stevenson M.Creutzfeld-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation Lancet Infect Dis 202020 e 2e 3187650410.1016/S 1473-3099(19)30615-2 · doi ↗ · pubmed ↗
- 3Kwon G.T.Kwon M.S.Diagnostic challenge of rapidly progressing sporadic Creutsfeldt-Jakob disease BMJ Case Rep 122019 e 23053510.1136/bcr-2019-230535 PMC 676834631551319 · doi ↗ · pubmed ↗
- 4Gao L.P.Tian T.T.Xiao K.Chen C.Zhou W.Liang D.L.Cao R.D.Shi Q.Dong X.P.Updated global epidemiology atlas of human prion diseases Front Public Health 122024141148910.3389/fpubh.2024.1411489 PMC 1120830738939567 · doi ↗ · pubmed ↗
- 5Rasheed U.Khan S.Khalid M.Noor A.Zafar S.A systemic analysis of Creutzfeldt Jakob disease cases in Asia Prion 181202411273832357410.1080/19336896.2024.2311950 PMC 10854368 · doi ↗ · pubmed ↗
- 6Zerr I.Kallenberg K.Summers D.M.Romero C.Taratuto A.Heinemann U.Breithaupt M.Varges D.Meissner B.Ladogana A.Schuur M.Haik S.Collins S.J.Jansen G.H.Stokin G.B.Pimentel J.Hewer E.Collie D.Smith P.Roberts H.Brandel J.P.van Duijn C.Pocchiari M.Begue C.Cras P.Will R.G.Sanchez-Juan P.Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease Brain 132Pt 102009265926681977335210.1093/brain/awp 191PMC 2759336 · doi ↗ · pubmed ↗
- 7Folstein M.Mc Hugh P.Mini mental state a practical method for grading cognitive state of patients for the clinician J Psychiatr Res 121975189198120220410.1016/0022-3956(75)90026-6 · doi ↗ · pubmed ↗
- 8Caine D.Nihat A.Crabb P.Rudge P.Cipolotti L.Collinge J.Mead S.The language disorder of prion disease is characteristic of a dynamic aphasia and is rarely an isolated clinical feature P Lo S One 1312018 e 019081810.1371/journal.pone.0190818 PMC 575588529304167 · doi ↗ · pubmed ↗