Methylation of mycovirus DNA is mediated by the RNAi machinery in vegetative hyphae of Fusarium graminearum
Yanfei Wang, Wei Chen, Lihang Zhang, Shuangchao Wang, Jin-Rong Xu, Lihua Guo

TL;DR
This study shows that DNA methylation in fungal hyphae helps defend against mycoviruses by silencing viral genes.
Contribution
The study reveals RNAi-mediated DNA methylation in vegetative hyphae of fungi as a novel antiviral defense mechanism.
Findings
DNA methylation of FgGMTV1 occurs in promoter regions, with up to 55.87% methylation in DNA-C fragment MeC5.
RNAi machinery components Ago and Dicer are essential for directing methylation of viral DNA and foreign promoters.
Deletion of RNAi genes leads to increased virus accumulation and impaired fungal growth and infection.
Abstract
DNA cytosine methylation is an important epigenetic mechanism for transposon silencing and gene regulation in fungi, particularly during sexual reproduction. However, its occurrence in vegetative hyphae and role in defense against mycoviruses is unclear. In this study, we demonstrated that genomic-wide cytosine methylation of the tripartite genomovirus FgGMTV1 occurs in the hyphae of Fusarium graminearum, a destructive pathogen of wheat and barley worldwide. Elevated methylation levels were predominantly observed in the promoter regions of FgGMTV1, with the highest level reaching 55.87% in the DNA-C fragment MeC5. Methylation of the Rep promoter in DNA-A was showed to be mediated by DNA methyltransferase DIM2 and lead to its transcriptional activity suppression, resulting in a significant reduction in virus accumulation. Furthermore, we uncovered that small RNAs (sRNAs) derived from…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
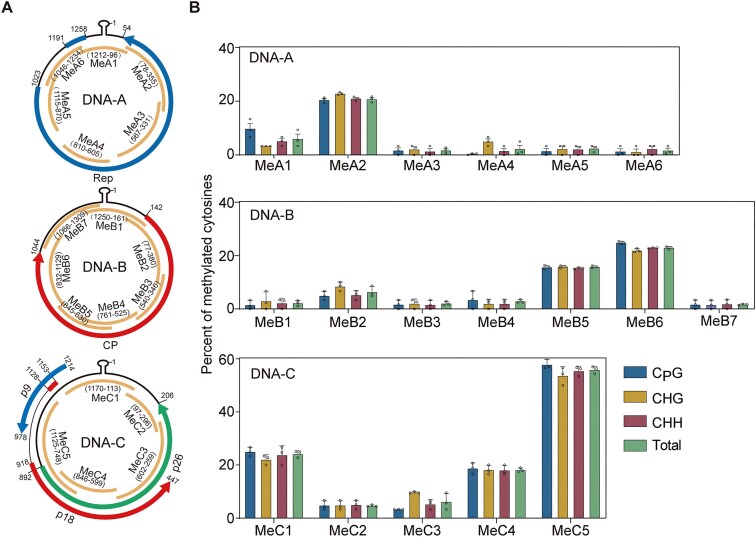 Figure 1
Figure 1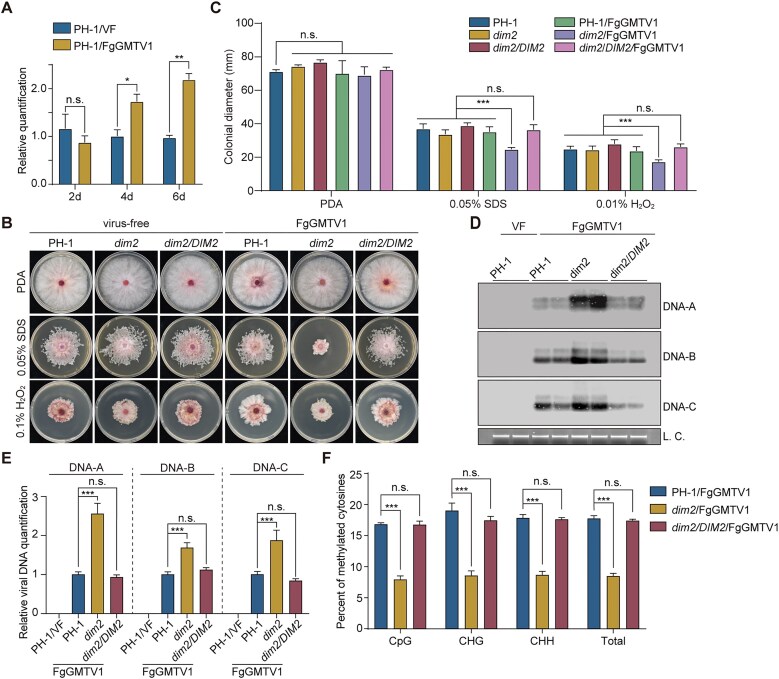 Figure 2
Figure 2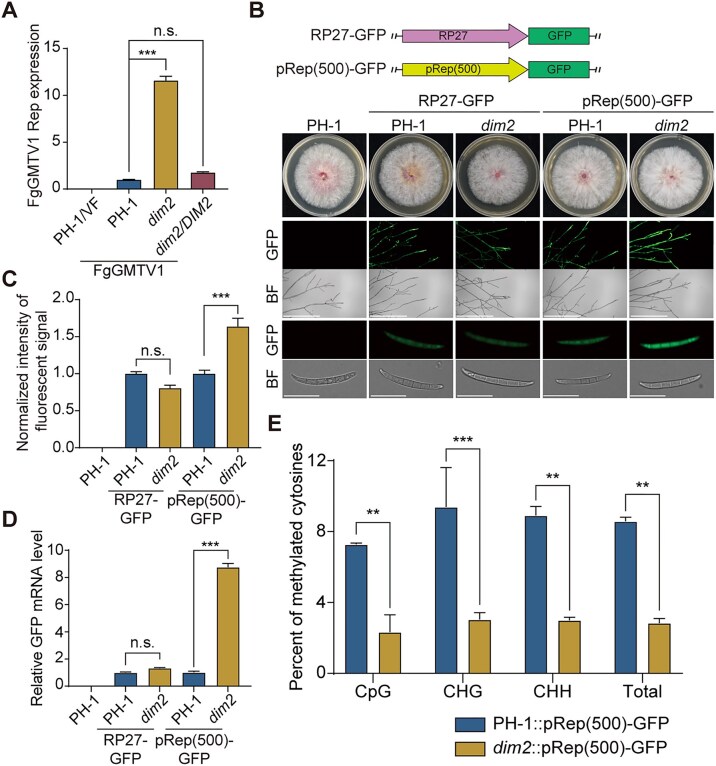 Figure 3
Figure 3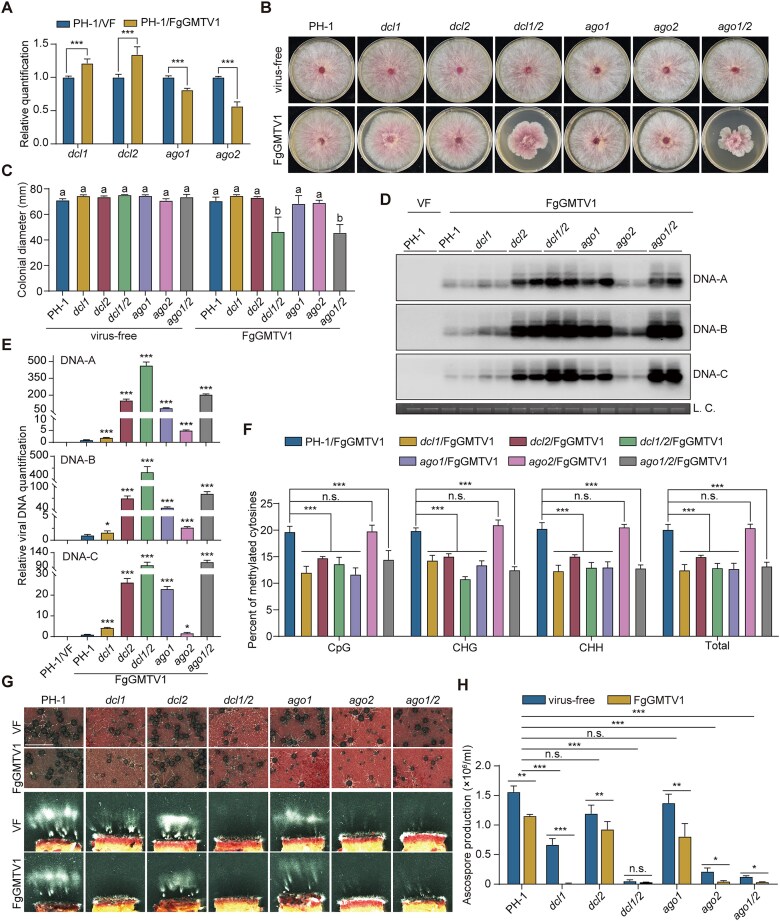 Figure 4
Figure 4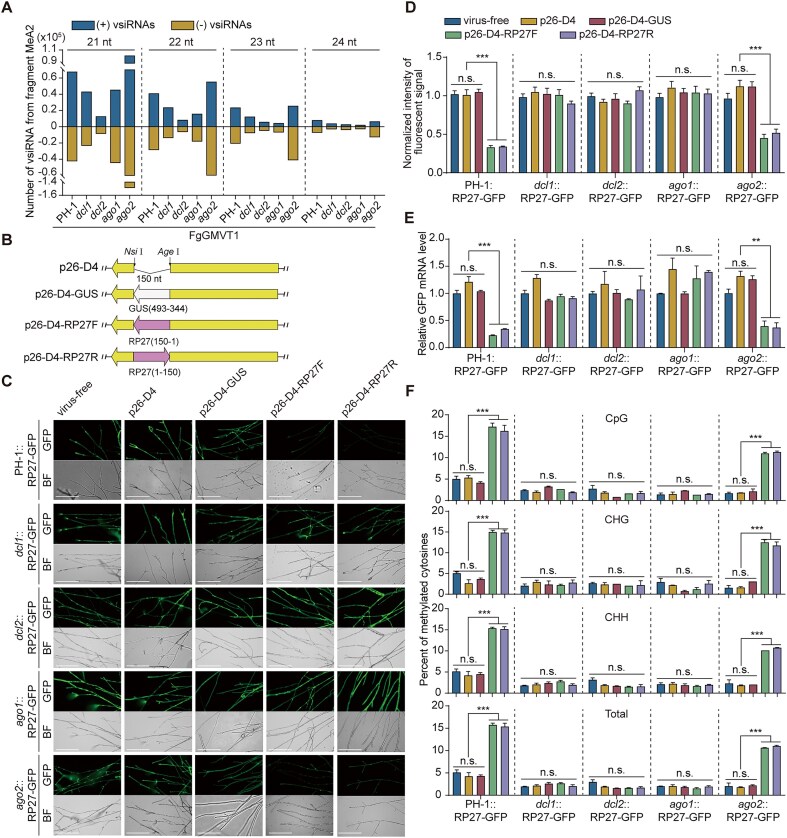 Figure 5
Figure 5- —National Natural Science Foundation of China10.13039/501100001809
- —Basic Research Center, Innovation Program of Chinese Academy of Agricultural Sciences
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Virus Research Studies · Plant and Fungal Interactions Research · Plant Disease Resistance and Genetics
Introduction
DNA cytosine methylation is a highly conserved epigenetic modification that plays pivotal roles in defending against nucleic acid invasions, maintaining genome integrity, and regulating gene expression in eukaryotes [1–3]. This modification involves the enzymatic conversion of cytosine bases to 5-methylcytosine by DNA methyltransferases (DNMTs). DNMTs are broadly categorized into two functional classes: “maintenance” methylation, which preserves methylation patterns post-DNA replication at hemimethylated sites, and “de novo” methylation, which introduces methylation at previously unmethylated cytosines [2]. In fungi, DNA methylation is predominantly associated with two genome defense mechanisms: repeat-induced point mutation (RIP) and methylation-induced premeiotically (MIP), both of which function during sexual reproduction [4, 5]. In the model filamentous ascomycete Neurospora crassa, DNA methylation occurs in transposons that have been rendered A:T-rich by the RIP defense system, a process facilitated by deficient in methylation-2 (DIM-2) and accompanied by histone H3K9 methylation [6–8]. Similarly, in Ascobolus immersus, DNA methylation specifically targets duplicated DNA sequences during MIP, with DNMT Masc1 acting as a key mediator prior to meiosis [9, 10]. Although DNMTs and genome-wide DNA methylation patterns have been characterized across diverse fungal species, these activities are predominantly observed in repetitive sequences during sexual reproduction [11, 12]. Comparatively, little is known about the occurrence and role of these methylation events during the vegetative cycle of fungi.
In plants and animals, viral DNA methylation serves as a critical defense mechanism against viral infections [13, 14]. For instance, when geminiviruses infect plants, their genomic DNA undergoes varying degrees of methylation [15]. The resistance genes Ty-1/Ty-3 in tomato, which encode the RNA-dependent polymerase RDRγ, confer resistance to tomato yellow leaf curl virus (TYLCV) through DNA methylation [16]. Transgenic plants expressing the β-Glucuronidase (GUS) reporter gene under the control of the tomato leaf curl virus (TLCV) promoter exhibit GUS silencing upon TLCV infection due to extensive promoter methylation [17]. These observations underscore the broad-spectrum defensive capabilities of DNA methylation in plants against geminiviruses. In animals, methylation of DNA tumor viruses serves to regulate the viral life cycle by influencing the transcription and expression of viral genes [18]. To circumvent this defensive, viruses have evolved suppressor proteins that disrupt DNA methylation or interfere with the biosynthesis of S-adenosyl-L-methionine (SAM), a crucial molecule in the DNA methylation pathway [15]. Despite its significance in other systems, DNA methylation of fungal viruses remains unexplored, likely due to the rarity of DNA mycoviruses in nature [19].
RNA interference (RNAi) is a conserved gene regulatory mechanism in eukaryotes including plants, animals, and fungi. It mediates post-transcriptional gene silencing (PTGS) by degrading or repressing target mRNAs and transcriptional gene silencing (TGS) by directing epigenetic modifications to the genome [20–22]. In plants, RNA-directed DNA methylation (RdDM) is the primary pathway for establishing de novo DNA methylation, relying on core RNAi machinery proteins such as Dicer-like (DCL) and Argonaute (AGO) [23]. In animals, PIWI-interacting RNAs (piRNAs) drive de novo methylation of differentially methylated regions (DMRs), independently of the RNAi pathway [24–26]. In Schizosaccharomyces pombe, small RNAs (sRNAs) induce TGS and co-transcriptional gene silencing (CTGS) through H3K9 methylation rather than DNA methylation [27]. Notably, Freitag et al. demonstrated that the RNAi pathway does not participate in the regulation of DNA methylation in N. crassa [28]. However, recent studies have revealed that DNA methylation in the human-pathogenic basidiomycete Cryptococcus neoformans is mediated by DNMT5, which contains sucrose nonfermenting (SNF2_N) and helicase_C domains homologous to those required for RdDM in plants [29–32]. Additionally, hypermethylation of transposable element (TE)-related sRNA clusters has been observed in basidiomycetous Pleurotus fungi [33]. Although these findings suggest the presence of an RdDM-like mechanism in the fungal kingdom, no direct evidence has been reported to date.
Fusarium graminearum gemytripvirus 1 (FgGMTV1), a member of the Genomoviridae family, is a tripartite circular single-stranded DNA mycovirus isolated from F. graminearum, an important pathogen causing Fusarium head blight (FHB) in cereal crops such as wheat, leading to significant yield loss and mycotoxin contamination [34, 35]. The first two genomic segments, DNA-A and DNA-B, encode a replication initiation protein (Rep) and a capsid protein (CP), respectively, while DNA-C encodes three proteins: p26, p9 (both with unknown functions), and p18, which facilitates asymptomatic viral infection in F. graminearum [36, 37]. The replication process of FgGMTV1, which is speculated to resemble that of geminiviruses, occurs in the nucleus via a rolling-circle mechanism, generating double-stranded DNA (dsDNA) intermediates that bind to host histones and form minichromosomes [38]. Small RNA sequencing has revealed that FgGMTV1-infected F. graminearum strains produce abundant viral siRNAs, suggesting activation of host RNA silencing mechanisms [39]. Moreover, a FgGMTV1-based virus-induced gene silencing (VIGS) system has been developed to attenuate hypervirulent F. graminearum strains, offering a potential strategy for FHB control [40].
In this study, we demonstrate that F. graminearum employs viral DNA methylation as a defense mechanism against FgGMTV1. Methylation of the Rep promoter in DNA-A, mediated by the DNMT DIM2, suppresses Rep transcriptional activity, thereby inhibiting virus accumulation. we further reveal a novel interplay between viral-derived sRNAs and the core RNAi proteins DCL1, DCL2, and AGO1, which collectively guide the methylation of both viral DNA and integrated foreign promoters. Notably, although DCL1 and AGO2 primarily mediate the sex-specific RNAi pathway in F. graminearum [41], our findings highlight functional redundancy within the DCL and AGO families, with DCL2 and AGO1 playing pivotal roles in the antiviral pathway. This study provides groundbreaking evidence that DNA methylation in fungi is dependent on RNAi, marking a significant advancement in fungal epigenetics. Furthermore, our results underscore the critical role of DNA methylation in fungal antiviral defense, shedding light on the complex dynamics of host-pathogen interactions.
Materials and methods
Fungal strains and culture conditions
The virus-free wild-type strain PH-1 of F. graminearum was used as the parent strain [42]. All fungal strains used in this study are listed in Supplementary Table S1. The strains were cultured in the potato dextrose agar (PDA) medium for 3–5 days at 25°C for morphological observation or in the cellophane-covered PDA medium for DNA and RNA extraction. For spore counting, the strains were cultured in the carboxymethyl cellulose (CMC) liquid medium for 5–7 days at 25°C to obtain the spores. For sexual reproduction experiments, 7-day-old aerial hyphae grown on carrot agar cultures (250 g/L) were pressed down using sterile 0.1% Tween-20 and subsequently cultured under black light at 25°C. The formation and development of perithecia, asci, ascospores, and ascospore discharge were examined as previously described [43, 44]. To count the number of ascospore discharges, the carrot agar was inverted for 12 h to allow mature perithecia to release ascospores onto the petri dish covers. The ascospores were then washed off the slide using 1 mL of sterile water, and their number was enumerated.
Strain and plasmid construction
Mutant alleles for targeted gene deletion were generated using double-joint (DJ) PCR, as previously described [45]. Briefly, 5′ and 3′ flanking regions of each gene were amplified using the primer pairs listed in Supplementary Table S1, and the amplified sequences were fused with the hygromycin resistance gene (HPH) cassette driven by the constitutive trpC promoter. Then PEG-mediated protoplast transformation of F. graminearum was performed, as previously described [40]. All putative gene-deletion mutants were identified via PCR using the relevant primers (Supplementary Table S1). F. graminearum dcl1, dcl2, and dcl1/2 mutants [46] were kindly provided Jin-Rong Xu, Purdue University, West Lafayette, Indiana, USA. To complement the dim2 deletion mutant (dim2), a fusion construct containing dim2 with its own promoter, neomycin resistance gene (NEO), and 3′-flanking region of dim2 was generated via DJ PCR. The fusion construct was further transformed into dim2 to generate the complementary strain, dim2/DIM2. The transformants were selected using geneticin (G418).
To generate the constructs to analyze the Rep promoter activity, 500-nt sequence upstream of the DNA-A-encoded Rep ATG was PCR-amplified and cloned between the Kpn I and BamH I sites of pGTN (RP27-GFP) to replace the RP27 promoter of GFP gene and generate pRep(500)-GFP using Seamless Cloning Kit (LABLEAD lnc., Beijing, China). The empty pGTN vector, RP27-GFP, or the pRep(500)-GFP vector were transformed into PH-1 and dim2 to generate PH-1::RP27-GFP, dim2::RP27-GFP, PH-1::pRep(500)-GFP, and dim2::pRep(500)-GFP.
The transfection of F. graminearum with the viral infectious clone pSK-ABC was performed using a PEG-mediated protoplast transfection method, as previously described [40].
To verify the effect of small RNA-mediated DNA methylation on RP27 promoter activity, we transformed the empty pGTN vector with RP27-GFP into PH-1, dcl1, dcl2, ago1, and ago2 strains to generate PH-1::RP27-GFP, dcl1::RP27-GFP, dcl2::RP27-GFP, ago1::RP27-GFP, and ago2::RP27-GFP strains, respectively. To construct VIGS vectors for the RP27 promoter, 150 bp sequences of RP27 promoter were cloned from the pGTN vector and inserted into the FgGMTV1 VIGS vector p26-D4 digested with Nsi I and Age I [35]. The 150-bp sequence of GUS gene was also cloned from the pCAMBIA1301 vector and inserted into the p26-D4 vector. Plasmids p26-D4-GUS, p26-D4-RP27F (sense), and p26-D4-RP27R (antisense) were also constructed. All primers used here are listed in Supplementary Table S2. The established VIGS vectors were transfected into PH-1::RP27-GFP, dcl1::RP27-GFP, dcl2::RP27-GFP, ago1::RP27-GFP, and ago2::RP27-GFP strains to obtain corresponding strains. Protoplast preparation and viral transfection were performed as previously described [35].
RNA extraction and RT-qPCR
RNA was extracted using the FastPure Universal Plant Total RNA Isolation Kit (Nanjing Vazyme Biotech, Nanjing, China) and reverse-transcribed into cDNA synthesis was carried out using Hifair III 1st Strand cDNA Synthesis Kit (Yeasen Biotechnolog Co. Ltd., Shanghai, China). Real-time qRT-PCR was performed using QuantStudio 5 (ABI) with Hieff qPCR SYBR Green MasterMix (Yeasen Biotechnology Co. Ltd., Shanghai, China). FgEF-1α was used as the internal control. All primer sequences are listed in Supplementary Table S2.
DNA extraction, southern blotting analysis, and qPCR
Total DNA was extracted from fungi using the cetyltrimethylammonium bromide method. Southern blotting was used to analyze viral DNA accumulation. Briefly, 1 μg of total DNA was separated on 1.5% agarose gels. After denaturation and neutralization of the agarose gels, total DNA was transferred to Hybond N + nylon membranes (Cytiva, Washington, USA). Hybridization and signal detection were performed using the DIG-High Prime DNA Labeling and Detection Starter Kit II (Roche, Basel, Switzerland). Three DNA probes, DNA-A, DNA-B, and DNA-C, were used to detect FgGMTV1, as previously described [36]. To detect viral DNA accumulation using qPCR, partial Rep fragments of 183 bp, partial CP fragments of 181 bp, and partial p26 fragments of 200 bp were amplified. All primer sequences used for qPCR are listed in Supplementary Table S2.
Responses of mutant strains to various stressors and pathogenicity assays
To determinate the responses of mutants to various stressors, mycelial plugs (5 mm in diameter) taken from the periphery of the 4-day-old colony of each strain were inoculated into the PDA medium mixed with hydrogen peroxide (0.1% (v/v) H_2_O_2_) and cell membrane-disrupting agents (0.05% (w/v) SDS), respectively. After the plates were incubated at 25°C for 4 days, colony diameter in each plate was measured. Each experiment was repeated three times. To assess the pathogenicity in the flowering wheat heads (cultivar Jimai22), a small equisized mycelial plug was inoculated in the glume of a spikelet and cultured in the field for 15 days. To detect virulence on seedling leaves (cultivar Jimai22), a 5-mm mycelial plug of each strain was inoculated into the center of the leave, and cultured at 25°C and 100% humidity with 12 h of daylight, and examined at 7 days. Each experiment was repeated three times.
Evaluation of GFP fluorescence
Sterile glass coverslips (20 × 20 mm) were placed on cellophane overlaying the PDA medium, and a small mycelial plug was inserted at the edge of the coverslip. When the mycelia grew on the coverslip, the coverslip with the attached mycelia was transferred to a glass slide, and GFP fluorescence was observed using a microscope (Carl Zeiss, Oberkochen, Germany). GFP fluorescence of spores, which were obtained by culturing the strains in the CMC liquid medium for 5–7 days at 25°C, was observed using a microscope (Carl Zeiss, Oberkochen, Germany). GFP fluorescence intensity was quantified using the ImageJ software [47].
DNA bisulfite sequencing analysis
Total DNA (500 ng) was subjected to bisulfite treatment using the DNA Bisulfite Transformation Kit (TIANGEN biotech CO. LTD., Beijing, China), according to the manufacturer's instructions. Bisulfite-treated DNA was used as the template for PCR amplification. The PCR products were ligated into the pMD18-T vector. Approximately 30 colonies were sequenced, and cytosine methylation was analyzed using the CyMATE program (http://cymate.org/cymate.html) [48]. Sequences with the same cytosine methylation status were counted as clones. At least three technical replicates of bisulfite treatment were performed for each viral infection experiment.
Virus-derived small RNA analysis
Mycelial masses were collected at the same time point after a 4-day culture period. Total RNA was extracted using the RNA Extraction Kit (TIANGEN Biotech CO. LTD., Beijing, China). RNA quality was evaluated with a BioAnalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). The cDNA libraries of sRNA were constructed with the QIAseq sRNA Library Kit (Qiagen, Hilden, Germany). In brief, 3′ and 5′ adapters were sequentially ligated to sRNAs, followed by unique molecular identifier (UMI)-based first-strand cDNA synthesis. Subsequent steps included cDNA purification and PCR amplification using a universal forward primer combined with index-containing reverse primers. Library quality control was performed using the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Sequencing was conducted on an Illumina NextSeq 6000 platform (Illumina, San Diego, CA, USA). Raw sequencing data were processed by removing low-quality reads and adapter contaminants using FASTX-Toolkit (v0.0.13) and filtering reads outside the 21–24 nt size range using Cutadapt (v4.1). The remaining reads with lengths of 21–24 nt were mapped to the genome of FgGMTV1 (GenBank accession: GCA_018594965.1) using the Bowtie software (http://bowtie-bio.sourceforge.net). The number of vsiRNA reads was normalized to reads per million (RPM) values (RPM = number of reads on a sRNA × 10^6^/total number of reads) according to the number of total reads in the corresponding small RNA library. The library construction and sequencing were performed at Shanghai Biotechnology Corporation.
Statistical analysis
Data are represented as the mean ± standard deviation (SD). Statistical analysis was performed using GraphPad Prism version 8.0. Significant differences between the control and treatment groups were analyzed using the one-way analysis of variance (ANOVA) followed by Dunnett's post-hoc test for pairwise comparisons. Statistical significance was set at P < 0.05 (*P < 0.05, **P < 0.01, and ***P < 0.001). The sample size (n) for each statistical analysis were reported in the corresponding “figure legends.”
Results
FgGMTV1 genome is a DNA methylation target
Methylation of viral genomes is an important epigenetic defense mechanism in animals and plants [13, 14]. To determine whether the FgGMTV1 genome is targeted by DNA methylation in F. graminearum, bisulfite sequencing was performed with the DNA-A, DNA-B, and DNA-C sequences of FgGMTV1 that were divided into six (MeA1–6), seven (MeB1–7), and five (MeC1–5) fragments, respectively (Fig. 1A). For DNA-A, fragment MeA2 had the highest methylation level (20.73%), with majority of methylation occurring at C_P_G, CHG, and CHH (Fig. 1B and Supplementary Fig. S1). The methylation level in other fragments of DNA-A range from 1.67% to 5.98%. For DNA-B, the fragments with the highest to lowest methylation levels were MeB6 (22.93%), MeB5 (15.79%), MeB2 (6.36%), MeB4 (2.86%), MeB1 (2.16%), MeB3 (1.92%), and MeB7 (1.66%). For DNA-C, fragment MeC5 had the average cytosine methylation level of 55.87%, with 57.88% of C_P_G, 53.67% of CHG, and 54.43% of CHH (Fig. 1B and Supplementary Fig. S1).
The genome-wide DNA methylation of FgGMTV1. (A) Three components of FgGMTV1 were depicted in circular forms, with six fragments of DNA-A (MeA1–6), seven fragments of DNA-B (MeB1–7) and five fragments of DNA-C (MeC1–5) used for bisulfite sequencing analysis indicated as orange lines. The blue arrow in DNA-A represents the open reading frame (ORF) of Rep. The red arrow in DNA-B represents the ORF of CP. The blue, red, and green arrows in DNA-C represent the ORFs of p9, p18, and p26, respectively. (B) Percentage of methylated cytosines in each fragment of FgGMTV1 determined by bisulfite sequencing with DNA isolated 4-day-old PDA cultures. For each fragment, 30 clones were sequenced to estimate the mean and standard deviation. Values are represented as the mean ± standard deviation (SD) (n = 3 independent experiments).
Through FgGMTV1 transcript mapping analysis, we observed that cytosine methylation patterns in the viral genome displayed preferential enrichment within or proximal to promoter regions. Specifically, three key methylated fragments were identified: MeA2 (78–355 nt, DNA-A), putatively located in the promoter region controlling expression of the DNA-A-encoded Rep protein; MeC5 (748–1125 nt, DNA-C), mapped to the promoter driving expression of the DNA-C-encoded p26 protein; and MeC1 (1170–113 nt, DNA-C), associated with the regulatory sequence governing p9/p18 expression (Fig. 1A) [36, 37]. These results provide direct evidence that FgGMTV1 is affected by cytosine methylation and that methylation is particularly enriched in the sequences within or near promoters, suggesting epigenetic regulation of viral gene expression.
DNMT DIM2 plays a critical role in the methylation of FgGMTV1
In plants, mammals, and fungi, DNA methylation is catalyzed by conserved DNMTs, using SAM as the methyl donor. Through homology searches using DNMTs of N. crassa, the following putative DNMT genes were identified in F. graminearum: dim2 (FGSG_10766) and rid (FGSG_08648) [49]. The rid acts as a pseudogene as it contains a UA^644^G premature stop codon that requires stage-specific editing to encode a full-length functional protein during sexual reproduction [50, 51]. Here, we analyzed the dim2 levels in both the virus-free and FgGMTV1-infected wild-type strain PH-1 using reverse transcription-quantitative polymerase chain reaction (RT-qPCR). The abundance of dim2 transcripts was significantly elevated in four-day cultures of PH-1/FgGMTV1 compared to that in the virus free culture (Fig. 2A). This result indicates that FgGMTV1 infection significantly alters the accumulation of the dim2 transcripts in F. graminearum.
*DIM2 plays a critical role in the methylation of FgGMTV1. (A) Expression analyses of dim2 in virus-free and FgGMTV1-infected strains on the PDA medium for 2, 4, and 6 days in the dark by RT-qPCR. The expression level of FgEF-1α was used as the internal control. (B) Colony morphologies of the virus-free (top) and FgGMTV1-infected (bottom) strains on PDA with or without 0.05% SDS and 0.1% H2O2 after incubation for 3 or 5 days. (C) colony diameters of the marked strains on PDA after incubation for 3 or 5 days. (D) Southern blots of genomic DNA isolated from 4-day-old PDA cultures in the dark were hybridized with probes A, B, and C that are specific for DNA-A, DNA-B, and DNA-C of FgGMTV1, respectively. LC, loading control to show equal amounts of genomic DNA in each lane. (E) The abundance of DNA-A, DNA-B, and DNA-C of FgGMTV1 in the same samples was assayed by qPCR. The FgEF-1α gene was used as the internal control. (F) Percentage of methylated cytosines in fragment MeA2 of FgGMTV1 in FgGMTV1-infected PH-1, dim2, and dim2/DIM2 strains determined by bisulfite sequencing. For A, C, E, and F, values are represented as the mean ± SD (n = 3 independent experiments). Statistical significance was determined using one-way ANOVA followed by Dunnett's post-hoc test. *, P < 0.05; **, P < 0.01; **, P < 0.001; and n.s., not significant.
To determine the function of DIM2 in FgGMTV1 infection, dim2 was knocked out using a homologous recombination strategy. Two independent dim2 deletion mutant strains were obtained, and gene deletion was confirmed via PCR (Supplementary Fig. S2A). We transformed protoplasts of dim2 mutants with a functional dim2 gene under the control of its native promoter to produce the dim2/DIM2 complementation strain (Supplementary Fig. S2B). The infectious clone of FgGMTV1 was then transfected into PH-1, dim2, and dim2/DIM2 strains via polyethylene glycol (PEG)-mediated protoplast transfection. Southern hybridization analysis confirmed that all transfectants were infective and verified the successful establishment of FgGMTV1-infected PH-1, dim2, and dim2/DIM2 (Fig. 2B and D). When quantified by southern blot and qPCR analyses, the abundance of FgGMTV1 was significantly higher in the dim2 mutant than in PH-1 and the dim2/DIM2 transformant (Fig. 2D and E). Bisulfite sequencing analyses with fragments MeA2, MeB6, and MeC5 showed that their methylation levels were significantly reduced in the dim2 mutant in comparison with PH-1 and dim2/DIM2 (Fig. 2F and Supplementary Fig. S2C–F). These results indicate that DIM2 plays a critical role in the methylation of FgGMTV1 in F. graminearum.
To characterize the function of DIM2-mediated FgGMTV1 methylation, we assessed mycelial growth, sexual/asexual reproduction, responses to stress, and virulence. Compared to PH-1 and dim2/DIM2, the FgGMTV1-infected dim2 showed no significant difference in colony morphology, mycelial growth rate, and conidiation (Fig. 2B and C, and Supplementary Fig. S3A). During sexual reproduction, all strains produced normal perithecia with mature asci and exhibited normal ascospore discharge (Supplementary Fig. S3B and C). This is similar to N. crassa but distinct from Cryphonectria parasitica [52].
The FgGMTV1-infected strains were also examined for changes in response to oxidative (H_2_O_2_) and cell membrane (sodium dodecyl sulfate [SDS]). The FgGMTV1-infected dim2 displayed an increased sensitivity to SDS and H_2_O_2_ (Fig. 2B and C). In infection assays, the virulence of the strain dim2/FgGMTV1 showed similar characteristics to the strain PH-1/FgGMTV1 in flowering wheat heads and wheat seedlings leaves (Supplementary Fig. S3D–F). Our findings further revealed that genome methylation of FgGMTV1 enhances the fungi's adaptability to abiotic stress.
DIM2-mediated methylation suppresses FgGMTV1 Rep promoter-dependent transcriptional activity
The promoter DNA methylation usually inhibits gene transcription in animals, plants, and fungi [3]. In the context of plant geminiviruses, the Rep protein holds a pivotal role in viral genome replication through the rolling circle replication mechanism [15]. Specifically, the MeA2 fragment is situated precisely 500 bp upstream of the DNA-A-encoded Rep open reading frame (ORF) (Rep promoter). When assayed by RT-qPCR, the expression of Rep in the FgGMTV1-infected dim2 mutant was significantly elevated compared to PH-1 and dim2/DIM2 (Fig. 3A), indicating a suppressive role of DIM2-mediated methylation. Thus, we hypothesized that DIM2 induces DNA hypermethylation of the Rep promoter, leading to Rep transcriptional inhibition, thereby inhibiting the accumulation of FgGMTV1.
*DIM2-mediated methylation of viral genome suppresses promoter-dependent transcriptional activities. (A) Analysis of the transcription of FgGMTV1-encoded Rep in FgGMTV1-infected PH-1, dim2, and dim2/DIM2 strains by RT-qPCR. (B) Schematic diagram of GFP expression driven by the FgGMTV1-encoded Rep promoter (pRep(500)). Colony morphology and green fluorescence intensity of mycelia and spores in PH-1 and dim2 strains transformed with RP27-GFP and pRep(500)-GFP. All strains were cultured in the PDA medium for mycelia and in the CMC medium for spores. Bar = 0.5 mm (Upper); bar = 25 μm (Lower). (C) Normalized intensity of fluorescent signal in PH-1 and dim2 strains transformed with RP27-GFP and pRep(500)-GFP. The fluorescence intensity was quantified using the ImageJ software, with those in PH-1::RP27-GFP and PH-1::pRep(500)-GFP strains set as 1.00. (D) Relative expression levels of GFP in 4-day-old PDA cultures of PH-1 and dim2 strains expressing RP27-GFP and pRep(500)-GFP assayed by qRT-PCR with EF-1α as the internal control. (E) Cytosine methylation in the Rep promoter in PH-1 and dim2 strains transformed with pRep(500)-GFP. Samples were collected as described in (C). For A, C, D, and E, values are represented as the mean ± SD (n = 3 independent experiments). Statistical significance was determined using one-way ANOVA followed by Dunnett's post-hoc test. **, P < 0.01; **, P < 0.001; and n.s., not significant.
To test this hypothesis, we cloned the 500-bp sequence upstream of the Rep ORF before the green fluorescent protein (GFP) reporter gene (pRep(500)-GFP) and transformed pRep(500)-GFP into the wild-type strain PH-1 (Fig. 3B). The GFP fluorescent signal was observed in all G418-resistant transformants with pRep(500)-GFP, suggesting that the 500-bp sequence upstream of the Rep ORF was functional (Fig. 3B). Then, we transformed the pRep(500)-GFP into the dim2 mutant strain. The ribosomal protein 27 (RP27) promoter with a GFP reporter (RP27-GFP) was used as a negative control. The GFP fluorescent signal assay shows that while the intensity of green fluorescence of RP27-GFP exhibited a similar level in both strains PH-1::RP27-GFP and dim2::RP27-GFP, that of pRep(500)-GFP was increased greatly in strain dim2::pRep(500)-GFP versus PH-1::pRep(500)-GFP (Fig. 3B and C). This result was further confirmed by RT-qPCR analysis, which showed that corresponding GFP mRNA levels in dim2::pRep(500)-GFP were significantly increased compared to PH-1::pRep(500)-GFP (Fig. 3D). Next, we performed bisulfite sequencing to detect the methylation of the Rep promoter at high resolution. The methylation level of the Rep promoter was significantly lower in dim2::pRep(500)-GFP than in PH-1::pRep(500)-GFP (Fig. 3E and Supplementary Fig. S4A).
To investigate whether FgGMTV1 infection enhances the methylation level of the integrated pRep(500)-GFP, we introduced the infectious FgGMTV1 clone into PH-1::pRep(500)-GFP and dim2::pRep(500)-GFP strains through PEG-mediated protoplast transfection (Supplementary Fig. S5A). Southern hybridization analysis confirmed successful viral infection in all transfectants (Supplementary Fig. S5B). Comparative analysis revealed a significant reduction in green fluorescence intensity between FgGMTV1-infected strains and their virus-free counterparts (Supplementary Fig. S5A), a finding corroborated by RT-qPCR showing corresponding decreases in GFP mRNA levels following viral infection (Supplementary Fig. S5C). Next, we performed bisulfite sequencing to detect the methylation of the Rep promoter in viral and nuclear genomes. Compared to FgGMTV1-infected PH-1::pRep(500)-GFP, the Rep promoter in viral genome showed lower methylation in FgGMTV1-infected dim2::pRep(500)-GFP (Supplementary Fig. S4B and S5D). Then, the methylation level of the Rep promoter in nuclear genome was significantly higher in FgGMTV1-infected PH-1::pRep(500)-GFP than in virus-free PH-1::pRep(500)-GFP, while dim2::pRep(500)-GFP strains maintained consistent methylation levels regardless of infection status (Supplementary Fig. S4C and S5E). These results suggest that DIM2-mediated methylation suppresses the FgGMTV1 Rep promoter-dependent transcriptional activity.
DCLs and AGOs are involved in the DNA methylation of FgGMTV1
F. graminearum contains two Dicer (DCL1 and DCL2) and two Argonaute (AGO1 and AGO2) proteins [53] that are orthologous to the DCLs and AGOs important for methylation and defense against DNA viruses in plants [13]. To investigate whether DCLs and AGOs are involved in FgGMTV1 infection in F. graminearum, the expression of dcl and ago genes was detected in both the virus-free and FgGMTV1-infected wild-type strain PH-1 by RT-qPCR. In the FgGMTV1-infected PH-1 strain, the expression of dcl1 and dcl2 was notably elevated compared to the virus-free PH-1 strain. In contrast, the expression of ago1 and ago2 exhibited a significant decrease (Fig. 4A).
*DCLs and AGOs are involved in FgGMTV1 DNA methylation. (A) Relative expression levels of dcls and agos in virus-free and FgGMTV1-infected strains assayed by qRT-PCR with RNA isolated from 4-day-old PDA cultures. (B) Colony morphologies of virus-free (top) and FgGMTV1-infected (bottom) mutant strains. (C) Colonial diameters of virus-free and FgGMTV1-infected mutant strains measured with 4-day-old PDA cultures. Mean and standard deviations were estimated with data from three independent replicates. Different letters indicate significant differences according to one-way ANOVA followed by Dunnett’s post-hoc test (P < 0.05). (D) Southern blots of genomic DNA isolated from 4-day-old PDA cultures in the dark were hybridized with probes A, B, and C that are specific for DNA-A, DNA-B, and DNA-C of FgGMTV1, respectively. LC, loading control to show equal amounts of genomic DNA in each lane. (E) The abundance of DNA-A, DNA-B, and DNA-C of FgGMTV1 in the same samples was assayed by qPCR. The FgEF-1α gene was used as the internal control. (F) Percentage of methylated cytosines in fragment MeA2 of FgGMTV1 in marked mutant strains determined by bisulfite sequencing. (G) Perithecium formation and ascospore discharge in mating cultures of the virus-free and FgGMTV1-infected marked mutant strains on carrot agar plates. Bar = 0.5 mm. (H) Number of the discharge ascospores produced by virus-free and FgGMTV1-infected marked mutant strains on carrot agar plates. For A, E, F, and H, values are represented as the mean ± SD (n = 3 independent experiments). Statistical significance was determined using one-way ANOVA followed by Dunnett’s post-hoc test. *, P < 0.05; **, P < 0.01; **, P < 0.001; and n.s., not significant.
To determine whether DCLs and AGOs are involved in DNA methylation of FgGMTV1, FgGMTV1 was introduced by the infectious clone of FgGMTV1 via PEG-mediated protoplast transfection into F. graminearum single gene deletion mutants (dcl1, dcl2, ago1, and ago2) and double-knockout mutants (dcl1/2 and ago1/2) (Fig. 4B and Supplementary Fig. S6A–C). Southern blotting and qPCR analysis showed that the accumulation of FgGMTV1 in all dcls and agos deletion mutants was markedly higher than that of the FgGMTV1-infected PH-1 (Fig. 4D and E). Meanwhile, the accumulation of FgGMTV1 in double knockout mutants was significantly greater than in single knockout mutants, indicating that while DCL1 and AGO2 play a primary role in the sex-specific RNAi pathway [41], both members of the DCL and AGO families exhibit functional redundancies in the anti-FgGMTV1 response. Notably, the abundance of FgGMTV1 DNA was significantly elevated in FgGMTV1-infected dcl2 and ago1 mutants compared to dcl1 and ago2 mutants, suggesting that DCL2 and AGO1 play a pivotal role in the anti-FgGMTV1 pathway. Bisulfite sequencing results demonstrated significantly lower methylation in fragment MeA2 in FgGMTV1-infected strains of dcl1, dcl2, dcl1/2, ago1, and ago1/2, compared to the PH-1/FgGMTV1 strain (Fig. 4F and Supplementary Fig. S6D). These findings suggest that DCL1, DCL2, and AGO1 are involved in FgGMTV1 genome methylation.
Then, we evaluated colony morphology, mycelial growth rate, the responses to stress, virulence, and sexual/asexual reproduction of virus-free and FgGMTV1-infected dcls and agos deletion mutants. Compared to FgGMTV1-infected PH-1, abnormal colony morphology and severe growth retardation were observed in FgGMTV1-infected dcl1/2 and ago1/2 mutants (Fig. 4B and C). In virus-free strains, the deletion mutants of dcls and agos exhibited no significant alterations in SDS conditions compared to PH-1. However, upon infection with FgGMTV1, most of these mutants, with the exception of ago2, displayed elevated sensitivity to SDS compared to PH-1 (Supplementary Fig. S7A and B). Both in virus-free and FgGMTV1-infected conditions, the deletion mutants of dcls and agos showed increased sensitivity to H_2_O_2_ compared to PH-1 (Supplementary Fig. S7A and B). Additionally, when infected with FgGMTV1, the dcl1, dcl1/2, ago1, and ago1/2 mutants exhibited further increased sensitivity to H_2_O_2_ compared to their virus-free counterparts (Supplementary Fig. S7A and B). In the virulence assay, the virulence of the FgGMTV1-infected dcl1/2, ago1, and ago1/2 strains was significantly reduced compared to the wild-type strain on flowering wheat heads (Supplementary Fig. S7C and D). The deletion mutants of dcls and agos, with or without FgGMTV1, exhibited normal conidiation (Supplementary Fig. S7E). For sexual reproduction, the dcl1, dcl1/2, ago1, ago2, and ago1/2 mutants formed markedly smaller perithecia, along with a decrease in ascospore formation and discharge (Fig. 4G and H). Furthermore, FgGMTV1-infection had an obvious effect on decreasing ascospore production in all mutants (Fig. 4G and H). These findings revealed that DCL1, DCL2, and AGO1 play crucial roles in the anti-FgGMTV1 pathway mediated by DNA methylation.
DCLs and AGOs are involved in the RNAi-directed DNA methylation pathway of FgGMTV1
In plants, DNA methylation is closely related to 21–24 nt sRNAs [54]. To investigate the relationship between FgGMTV1 DNA methylation and siRNAs produced by the RNAi machinery, we analyzed the association between the distribution of virus-derived siRNAs (vsiRNAs) and DNA methylation in the FgGMTV1 genome [39]. An enrichment hotspot of FgGMTV1-derived siRNAs was observed in the MeA2, MeB6, and MeC5 DNA methylation hotspot regions (Supplementary Fig. S8A–C). Total vsiRNAs were subjected to deep sequencing analysis in the FgGMTV1-infected PH-1, dcl1, dcl2, ago1, and ago2 strains. Next, we analyzed the 21–24 nt vsiRNAs derived from the fragment MeA2 in all strains separately. The accumulation of 21–24 nt vsiRNAs derived from the fragment MeA2 in the FgGMTV1-infected dcl1, dcl2, and ago1 mutants was significantly decreased respectively, compared to that in PH-1/FgGMTV1, but that in FgGMTV1-infected ago2 was increased (Fig. 5A). Meanwhile, the 21–24 nt vsiRNAs derived from the fragment MeC5 showed a similar trend to that from the fragment MeA2 in the FgGMTV1-infected strains of dcl1, dcl2, and ago1 (Supplementary Fig. S8D). This observation suggests that the low DNA methylation level of FgGMTV1-infected strains of dcl1, dcl2, and ago1 in F. graminearum is correlated with a lower production of viral 21–24 nt siRNAs.
*DCLs and AGOs are involved in the RNA-directed DNA methylation pathway of fungi. (A) Normalized read number of 21–24 nt vsiRNAs derived from fragment MeA2 in sRNA libraries of the FgGMTV1-infected mutant strains. “(−)” and “(+)” indicate the sRNAs derived from the complementary (negative) and positive viral genomic strands, respectively. (B) The p26 regions in the empty p26-D4 vector and p26-D4 with fragments of GUS and RP27 promoter sequences closed in its multiple cloning site (MCS). Yellow, white, and pink arrows indicate the orientations of p26, GUS, and RP27 promoter sequences, respectively. Numbers indicate the location of GUS and the RP27 promoter. (C) Representative images of marked strains that are virus free or infected with labeled viral constructs infected with no p26-D4-RP27F/R-infected mutant strains. Bar = 200 μm. (D) Normalized intensity of fluorescent signal in p26-D4-RP27F/R-infected mutant strains. The fluorescence intensity was quantified using the ImageJ software, with those in virus free strains set as 1.00. (E) Relative levels of GFP mRNA in p26-D4-RP27F/R-infected mutant strains. RT-qPCR was performed using the FgEF-1α transcript levels as the internal control. (F) Percentage of methylated cytosines in the RP27 promoter of p26-D4-RP27F/R-infected mutant strains determined via bisulfite sequencing. For D, E, and F, bars of the same color represent the same strain samples. values are represented as the mean ± SD (n = 3 independent experiments). Statistical significance was determined using one-way ANOVA followed by Dunnett’s post-hoc test. **, P < 0.01; **, P < 0.001; and n.s., not significant.
In plants, RdDM can be manipulated to target and silence some genes in a highly specific manner. For example, foreign genes expressed under the control of the cauliflower mosaic virus (CaMV) 35S promoter are silenced by inducing hypermethylation of the 35S promoter following infection by a virus carrying the same promoter sequence [55, 56]. To investigate whether there is a similar phenomenon in fungi, we used the FgGMTV1-based VIGS vector p26-D4 [35] to construct VIGS vectors of the RP27 promoter, p26-D4-RP27F and p26-D4-RP27R, which carried 150 bp sense and antisense sequences from the RP27 promoter (Fig. 5B). These vectors p26-D4 (empty VIGS vector), p26-D4-GUS (as a VIGS control), p26-D4-RP27F/R were transfected into the PH-1::RP27-GFP strain via PEG-mediated protoplast transfection (Supplementary Fig. S9A). Southern blotting analysis revealed that all the vectors were infective (Supplementary Fig. S9B). To verify the efficacy of gene silencing, we assessed the fluorescence intensity and transcriptional levels of GFP in p26-D4-RP27F/R infected PH-1::RP27-GFP strains. Compared with that in the virus-free, p26-D4, and p26-D4-GUS-infected PH-1::RP27-GFP strains, green fluorescence intensity was significantly reduced in the p26-D4-RP27F/R-infected PH-1::RP27-GFP strains (Fig. 5C and D). This result was further confirmed via RT-qPCR analysis, which showed that corresponding GFP mRNA levels were lower in the p26-D4-RP27F/R-infected PH-1::RP27-GFP strains than in the virus-free, p26-D4, and p26-D4-GUS-infected strains (Fig. 5E).
We investigated the methylation status of the RP27 promoter in p26-D4-RP27F/R-infected PH-1::RP27-GFP strains. Bisulfite sequencing analyzes revealed that the methylation level of the RP27 promoter was significantly higher in the p26-D4-RP27F/R-infected PH-1::RP27-GFP strains than in the virus-free, p26-D4, and p26-D4-GUS-infected strains (Fig. 5F and Supplementary Fig. S10). These findings revealed that small RNA from FgGMTV1 mediated DNA methylation suppresses integrated RP27 promoter-dependent transcriptional activity.
To test the role of DCLs and AGOs in the RNAi-directed DNA methylation pathway, we generated integrated RP27-GFP expressing strains of dcl1::RP27-GFP, dcl2::RP27-GFP, ago1::RP27-GFP, and ago2::RP27-GFP, and transfected p26-D4-RP27F/R into them (Supplementary Fig. S9A). Southern blot analysis indicated that all the vectors were infective (Supplementary Fig. S9B). The fluorescence intensity analysis demonstrated that no reduction of GFP fluorescence intensity was observed in the p26-D4-RP27F/R-infected dcl1::RP27-GFP, dcl2::RP27-GFP, and ago1::RP27-GFP strains, compared to the virus-free, p26-D4, and p26-D4-GUS-infected strains (Fig. 5C and D). The relative level of GFP mRNA in the p26-D4-RP27F/R-infected dcl1::RP27-GFP, dcl2::RP27-GFP, and ago1::RP27-GFP strains are similar for the virus-free, p26-D4, and p26-D4-GUS-infected strains (Fig. 5E). However, similar to the p26-D4-RP27F/R infected PH-1::RP27-GFP strains, the GFP fluorescence intensity and relative level of GFP mRNA in the p26-D4-RP27F/R-infected ago2::RP27-GFP strains were significantly reduced (Fig. 5C–E). The hypomethylation of the RP27 promoter in p26-D4-RP27F/R-infected dcl1::RP27-GFP, dcl2::RP27-GFP, and ago1::RP27-GFP strains were observed, and was similar to the virus-free, p26-D4, and p26-D4-GUS-infected strains (Fig. 5F and Supplementary Fig. S10). But the methylation level of the RP27 promoter was significantly higher in p26-D4-RP27F/R infected ago2::RP27-GFP strains than in virus-free, p26-D4 or p26-D4-GUS infected strains (Fig. 5F and Supplementary Fig. S10). Taken together, these results suggest that DCL1, DCL2, and AGO1 are involved in the RNAi-directed DNA methylation pathway in fungi.
Discussion
Evolutionary conservation of viral promoter methylation in eukaryotic antiviral defense
DNA methylation-mediated TGS is a well-characterized defense mechanism against geminiviruses in plants [13]. These viruses replicate through dsDNA intermediates that assemble into nucleosomes, rendering them ideal substrates for methylation [38]. For example, mutants of Arabidopsis with deficient DNA methylation (e.g. cmt3, drm2, and ago4) exhibit increased sensitivity to CaMV and bean common mosaic virus, accompanied by reduced viral genome methylation [13]. Our findings extend this paradigm to the fungal kingdom through the characterization of F. graminearum’s DIM2-dependent methylation system targeting mycovirus FgGMTV1 (Fig. 2). Bisulfite sequencing revealed hypermethylation hotspots within viral promoter regions—specifically fragment MeA2 (78–355 nt) in DNA-A (Rep promoter), MeC5 (748–1125 nt) in DNA-C (p26 promoter), and MeC1 (1170–113 nt) governing p9/p18 expression [37] (Fig. 1). These findings mirror observations in Nicotiana benthamiana infected with tomato yellow leaf curl China virus (TYLCCNV), where promoter-adjacent hypermethylation occurs [57], suggesting promoter-targeted DNA methylation represents a universal eukaryotic defense mechanism against DNA viruses.
Viral promoter methylation typically suppresses transcription by either blocking transcription factor binding or inducing chromatin modifications [58, 59]. TLCV has been demonstrated to induce TGS of a homologous integrated plant transgene, which is characterized by extensive hypermethylation- of the virus-derived sequences within the transgenic locus [17]. To dissect this mechanism, we engineered transgenic strains with GFP expression driven by the Rep promoter. dim2 deletion reduced promoter methylation while increasing GFP expression (Fig. 3). Conversely, introducing p26-D4-RP27 triggered RP27 promoter hypermethylation and GFP silencing (Fig. 5). These results demonstrate that fungal viral infections generate sequence-specific methylation signals inducing TGS. Although DNA methylation is tightly linked to transcriptional repression, the underlying molecular machinery remains elusive in fungi. In plants, methyl-binding proteins (MBPs) recruit repressive complexes (e.g. HDACs, polycomb proteins) through distinct domains (MBD or Kaiso/BTB zinc fingers) [60, 61]. The absence of fungal MBP homologs suggests divergent silencing mechanisms, establishing our system as a unique platform to explore fungal-specific TGS pathways.
RdDM as an ancient and conserved regulatory strategy across eukaryotes
While RNAi-mediated DNA methylation was first described in plants as early as 1994 [62, 63], no fungal counterparts had been reported until now. Our work provides the first evidence of RdDM in fungi, resolving longstanding questions about RNAi-DNA methylation crosstalk in this kingdom. Plant canonical RdDM involves Pol IV/RDR2-dependent dsRNA synthesis, DCL3 processing into 24-nt sRNAs, and AGO4/6/9-guided DRM2 recruitment [23]. While Pol IV and Pol V are plant-specific multi-subunit RNA polymerases, they both evolved from Pol II, which is shared by eukaryotes [64, 65]. Pol II is supposed to serve as an enzyme assisting in the targeting of RdDM or RNA-directed histone modifications in invertebrates and yeasts, which lack specialised RdDM polymerases [66]. Intriguingly, FgGMTV1-derived vsiRNAs (21–22 nt) correlated spatially with methylation hotspots, with minimal 24-nt species detected (Fig. 5 and S7). This contrasts with canonical plant RdDM but aligns with non-canonical RdDM-associated pathways [23]. These findings not only illuminate fundamental epigenetic mechanisms in fungi but also imply that RNAi-directed DNA methylation may represent an ancient and conserved regulatory strategy across eukaryotes.
The functional repertoire of fungal DNMTs and RNAi components varies considerably across species. The fungal kingdom exhibits remarkable diversity in DNMTs, with major genotypes including DNMT1 + DNMT5, DIM2 + DNMT5 + RID, and DNMT1 [12]. While RNAi pathways are essential for regulating vegetative growth and conidiation in Cryptococcus neoformans, N. crassa, Mucor circinelloides, and Magnaporthe oryzae [67–70], F. graminearum RNAi-deficient mutants (dcl1/2 and ago1/2) exhibit no discernible defects in these developmental processes (Fig. 4). In addition, RNAi is essential for antiviral defense in C. parasitica [71] and F. graminearum (Fig. 4). Given the diversity of DCLs, AGOs, and DNMTs across fungi, further research is needed to elucidate the conservation and divergence of RNAi-mediated DNA methylation in other fungal species.
Integrated antiviral strategies: balancing immediate and sustained responses
Our dissection of F. graminearum’s defense architecture reveals a sophisticated layering of antiviral mechanisms. DCL2 and AGO1 play dual roles in antiviral defense mechanisms, contributing not only to RNAi-mediated DNA methylation but also to PTGS in F. graminearum [72, 73]. Our study reveals that dim2 deletion exerts a more pronounced negative effect on FgGMTV1 Rep promoter methylation compared to disruption of key RNAi components (Fig. 2F and 4F). Notably, while dim2 knockout only moderately increases viral load (approximately 2-fold), RNAi mutations result in substantially greater viral accumulation (>10-fold), indicating that PTGS functions as the principal antiviral defense mechanism, with RdDM operating as a secondary regulatory strategy. This functional hierarchy resembles transitional models proposed in plants, where RNAi signals bridge acute PTGS responses to persistent TGS states [23].
Notably, residual DNA methylation persists in mutants lacking dim2, dcls, or agos, suggesting the existence of methylation pathways independent of both DIM2 and the RNAi machinery. This observation parallels findings in N. crassa, where DNA methylation is associated with RNAi-independent processes such as RID-mediated RIP and DIM5-dependent H3K9 methylation [6, 7]. These collective findings propose that F. graminearum likely harbors an alternative RNAi-independent DNA methylation pathway—a hypothesis demanding systematic investigation.
Conclusion and perspectives
Beyond advancing fundamental mycovirology, our findings establish a pioneering framework for RNA-guided epigenetic therapeutics. While current RNA-based approaches predominantly focus on targeting RNA molecules through antisense oligonucleotides or RNAi triggers [74], we demonstrate a paradigm-shifting capability: sequence-specific sRNAs can direct locus-specific DNA methylation via RdDM mechanisms in fungi. This breakthrough provides proof-of-concept for engineering synthetic “epigenetic RNAs” capable of programming precise DNA methylation patterns, opening transformative avenues including the development of sequence-specific DNA methyltransferases guided by designer RNAs, the engineering of cross-kingdom epigenetic regulation systems, and the creation of RNA-based chromatin editors with broad agricultural and biomedical applications. Future investigations should prioritize resolving key mechanistic questions, including the functional role of RNA polymerase II in fungal RdDM compared to plant-specific Pol IV/V systems, and structural determinants governing DIM2 targeting by AGO1-vsiRNA complexes. Given this phylogenetic conservation of RdDM across plants and fungi, intriguing questions arise about its potential role in animals and humans. Addressing these knowledge gaps will be essential for translating this epigenetic programming platform into practical technologies.
In summary, by establishing F. graminearum as a model for fungal epigenetic virology, this work bridges critical gaps in understanding eukaryotic antiviral strategies. The discovery of RNAi-dependent DNA methylation challenges existing paradigms of fungal epigenetics while revealing deep evolutionary connections across eukaryotes. These insights advance our understanding of host-virus coevolution and catalyze innovations in RNA-based epigenetic engineering.
Supplementary Material
gkaf478_Supplemental_File
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Martienssen RA, Colot V DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science. 2001; 293:1070–4.10.1126/science.293.5532.1070.11498574 · doi ↗ · pubmed ↗
- 2Zhang HM, Lang ZB, Zhu JK Dynamics and function of DNA methylation in plants. Nat Rev Mol Cell Biol. 2018; 19:489–506.10.1038/s 41580-018-0016-z.29784956 · doi ↗ · pubmed ↗
- 3Schubeler D Function and information content of DNA methylation. Nature. 2015; 517:321–6.10.1038/nature 14192.25592537 · doi ↗ · pubmed ↗
- 4Madhani HD Unbelievable but true: epigenetics and chromatin in fungi. Trends Genet. 2021; 37:12–20.10.1016/j.tig.2020.09.016.33092902 PMC 8994648 · doi ↗ · pubmed ↗
- 5Miao VP, Freitag M, Selker EU Short Tp A-rich segments of the ζ-η region induce DNA methylation in Neurospora crassa. J Mol Biol. 2000; 300:249–73.10.1006/jmbi.2000.3864.10873464 · doi ↗ · pubmed ↗
- 6Tamaru H, Selker EU A histone H 3 methyltransferase controls DNA methylation in Neurospora crassa. Nature. 2001; 414:277–83.10.1038/35104508.11713521 · doi ↗ · pubmed ↗
- 7Freitag M, Williams RL, Kothe GO et al. A cytosine methyltransferase homologue is essential for repeat-induced point mutation in Neurospora crassa. Proc Natl Acad Sci USA. 2002; 99:8802–7.10.1073/pnas.132212899.12072568 PMC 124379 · doi ↗ · pubmed ↗
- 8Kouzminova E, Selker EU dim-2 encodes a DNA methyltransferase responsible for all known cytosine methylation in Neurospora. EMBO J. 2001; 20:4309–23.10.1093/emboj/20.15.4309.11483533 PMC 149169 · doi ↗ · pubmed ↗