Immunoglobulin M Nephropathy: A Diagnostic Dilemma Between Minimal Change Disease and Focal Segmental Glomerulosclerosis
Gautam Agrawal, Bhawna Agarwal, Kunal Sonavane, Pallavi Shirsat

TL;DR
This paper discusses the challenges in diagnosing IgM nephropathy, a kidney disease that can be mistaken for other conditions, and highlights the need for better treatment guidelines.
Contribution
The paper emphasizes IgMN as a distinct diagnosis and highlights the need for standardized treatment protocols.
Findings
IgMN shares features with MCD and FSGS but is a distinct clinicopathological diagnosis.
Patients with IgMN are at risk of progressing to end-stage renal failure.
Corticosteroids, calcineurin inhibitors, and rituximab are potential treatment options.
Abstract
Immunoglobulin M nephropathy (IgMN) is an idiopathic glomerulonephritis characterized by diffuse IgM deposits in the mesangium and mesangial hypercellularity. It has been a controversial diagnosis since it was initially described, as IgMN shares features with both minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS). However, studies have shown that IgMN is a distinct clinicopathological diagnosis comprising patients with predominant mesangial IgM deposits and who do not meet established criteria for MCD and FSGS. We present a case of a 24-year-old male presenting with hypertension and proteinuria, with kidney biopsy showing concern for IgMN. These patients carry a significant risk of developing nephrotic syndrome, renal insufficiency, and progression to end-stage renal failure. This underscores the importance of timely diagnosis of IgMN and therapeutic…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
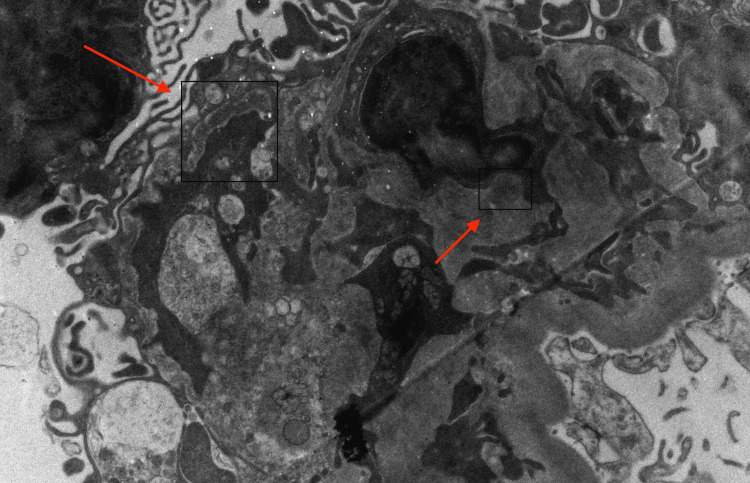 Figure 1
Figure 1| Laboratory tests | Results | Reference range |
| 24-hour protein | 248 | <150 mg |
| Serum albumin | 4.6 | 3.5-5.7 gm/dl |
| Blood urea nitrogen (BUN) | 17 | 7-25 mg/dl |
| Creatinine | 1.3 | 0.6-1.2 mg/dl |
| Calcium | 9.8 | 8.6-10.3 mg/dl |
| Hemoglobin | 15.6 | 11.7-15.8 gm/dl |
| ANA titer | Negative | Negative |
| Anti-dsDNA | Negative | <25 - negative |
| Hemoglobin A1c | 5.5 | 5.7-6.4 (prediabetic) % |
| Hepatitis B surface antigen | Non-reactive | Non-reactive |
| Hepatitis C antibody | Non-reactive | Non-reactive |
| Category | IgM nephropathy | Minimal change disease (MCD) | Focal segmental glomerulosclerosis (FSGS) |
| Clinical presentation | Nephrotic syndrome, non-nephrotic range proteinuria, or hematuria | Nephrotic syndrome | Nephrotic syndrome with hypertension and renal insufficiency |
| Biopsy findings | Diffuse IgM deposits in mesangium and mesangial proliferation | Normal glomeruli on light microscopy, effacement of podocyte foot processes on electron microscopy | Sclerosis affecting a portion of the glomerulus, effacement of the podocyte foot process |
| Response to treatment | Variable response to steroids, higher rate of steroid dependence and resistance as compared to MCD | High response rate to steroids | Poor response rate to steroids, often requires other immunosuppressants |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRenal Diseases and Glomerulopathies · Amyloidosis: Diagnosis, Treatment, Outcomes · Chronic Kidney Disease and Diabetes
Introduction
Immunoglobulin M nephropathy (IgMN) is an idiopathic glomerulonephritis characterized by mesangial hypercellularity and diffuse mesangial deposition of IgM [1]. The reported frequency of IgMN has been variable, ranging from 1.8% to 18.5% in native kidney biopsies [2]. It has remained a controversial diagnosis since it was first described in the 1970s, as IgMN shares features with both minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS) [2]. IgMN is considered to fall in the spectrum between MCD and FSGS, but it is evident that there is a distinct group of patients, identifiable by immunohistochemistry, who exhibit predominant mesangial IgM deposition and do not meet the established diagnostic criteria for either MCD or FSGS [2]. Clinically, IgMN can present with nephrotic syndrome, but it also shows more mesangial proliferation and matrix expansion, which are not typical of MCD. Histologically, IgMN exhibits more tubular atrophy and interstitial fibrosis compared to MCD, aligning it closer to FSGS [3].
The Mayo Clinic/Renal Pathology Society provides specific guidelines for diagnosing IgMN. Typically, IgMN presents mesangial hypercellularity and diffuse mesangial IgM deposits, but IgMN can be associated with tubular atrophy and interstitial fibrosis, which are significant predictors of renal insufficiency and considered to be poor prognostic markers [4].
We present a case of a 24-year-old male who presented with hypertension, found to have proteinuria, and further evaluation with kidney biopsy showed concern for IgMN.
Case presentation
A 24-year-old healthy male was referred to the nephrology clinic after being found to have hypertension. He denied any significant past medical history, including chronic kidney disease. He was born at full term. His father had a notable history of hypertension and congestive heart failure and died at the age of 50 from an unknown cause. The patient denied any known family history of chronic kidney disease.
On initial examination, the patient weighed 232 lbs. His blood pressure was 130/84 mmHg in the left upper arm. He appeared well-built, alert, and oriented. Pulmonary examination revealed clear breath sounds bilaterally, with no crackles or wheezes. Cardiac examination revealed regular heart sounds. There was no peripheral edema, rash, or tattoos noted.
Initial laboratory workup revealed a serum creatinine of 1.3 mg/dL, with an estimated glomerular filtration rate (eGFR) of 68 mL/min/1.73 m². Urinalysis showed trace proteinuria and microscopic hematuria. A 24-hour urine collection revealed proteinuria of 248 mg/day. Serologic testing was negative for antinuclear antibodies and double-stranded DNA antibodies, with normal complement levels. Other laboratory results were unremarkable (Table 1).
The patient underwent a kidney biopsy, which revealed minimal histopathological changes with mildly increased mesangial cellularity and weak mesangial IgM, C3, and kappa-dominant deposits. Electron microscopy showed 15% podocyte foot process effacement and equivocal mesangial electron-dense deposits (Figure 1), with focal endothelial hypertrophy and questionable basement membrane duplication. No chronic changes, glomerulosclerosis, or evidence of arterionephrosclerosis, interstitial fibrosis, or tubular atrophy were observed. These findings were concerning for IgM nephropathy.
Kidney biopsy image.Red arrows showing mesangial deposits.
He was initiated on aggressive blood pressure control with lisinopril, later supplemented with amlodipine, resulting in improvement in both blood pressure and proteinuria. His proteinuria has improved to 154 mg in the most recent 24-hour urine collection. The current management plan involves continued monitoring of proteinuria, with consideration of corticosteroid therapy in the event of future flares.
Discussion
Our patient presented with hypertension at a young age, with further evaluation showing proteinuria, which necessitated further evaluation with kidney biopsy, and the diagnosis of IgMN was established. We would like to highlight the importance of appropriate diagnostic evaluation for patients with hypertension at a young age and proteinuria with renal biopsy, given that timely therapeutic intervention in these patients is of utmost importance to prevent kidney function decline.
IgM nephropathy can present as sub-nephrotic range proteinuria, hematuria, or nephrotic syndrome, which is commonly characterized by steroid dependence or resistance, with a significant risk of progression to end-stage renal disease. IgMN shares clinical and histopathological features with MCD and FSGS [4]. Key differences between IgM nephropathy, MCD, and FSGS are outlined in Table 2.
In a study, when IgMN was divided into subtypes, patients with FSGS-like IgMN had lower eGFR and higher proteinuria compared to those with MCD-like IgMN. Diagnosis of hypertension at the time of kidney biopsy was identified as a predictor of a ≥20% decline in eGFR over two years in IgMN patients [5]. Investigators suggest that repeat biopsies in patients with IgMN often show FSGS despite ongoing mesangial IgM deposition [2]. Researchers have shown that over 15 years, 36% of patients with IgMN developed renal insufficiency, and 23% progressed to end-stage renal failure (ESRD). Interstitial fibrosis was found to have the strongest prognostic value among histological parameters [1].
Renin-angiotensin system inhibitors are recommended as part of the standard treatment for IgMN. Treatment options also include corticosteroids or calcineurin inhibitors for nephrotic range proteinuria, but steroid dependence and resistance are higher for IgMN as compared to MCD. Research has shown that among IgMN patients with nephrotic syndrome, 29% were steroid-resistant, while 80% of those who initially responded to steroids became steroid-dependent [1]. Rituximab has also demonstrated efficacy in inducing remission in refractory cases of IgMN, making it a valuable therapeutic option. Rituximab has shown efficacy in treating steroid-dependent or frequently relapsing nephrotic syndrome in MCD and FSGS, reducing relapse rates and steroid dependence. This aligns with the successful use of rituximab in the IgMN case [6,7]. Further research and clinical trials are needed to establish its role more definitively in the management of IgMN.
Conclusions
This case underscores the critical importance of thorough diagnostic evaluation in young patients presenting with hypertension and proteinuria. Early consideration of renal biopsy can lead to timely diagnosis of underlying glomerular diseases such as IgM nephropathy. Prompt identification and appropriate management are essential to prevent progression of renal dysfunction and to optimize long-term outcomes. There is a need for robust clinical trials on IgMN to establish standardized treatment protocols and long-term outcomes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ig M nephropathy: clinical picture and long-term prognosis Am J Kidney Dis Myllymäki J Saha H Mustonen J Helin H Pasternack A 343350412003 https://www.ajkd.org/article/S 0272-6386(02)69144-9/abstract 1255249510.1053/ajkd.2003.50042 · doi ↗ · pubmed ↗
- 2The natural history of immunoglobulin M nephropathy in adults Nephrol Dial Transplant Connor TM Aiello V Griffith M Cairns T Roufosse CA Cook HT Pusey CD 823829322017 https://doi.org/10.1093/ndt/gfw 0632719037910.1093/ndt/gfw 063PMC 5837784 · doi ↗ · pubmed ↗
- 3Ig M nephropathy: is it closer to minimal change disease or to focal segmental glomerulosclerosis?J Nephrol Brugnano R Del Sordo R Covarelli C Gnappi E Pasquali S 479486292016 https://doi.org/10.1007/s 40620-016-0269-62684262410.1007/s 40620-016-0269-6 · doi ↗ · pubmed ↗
- 4Mayo Clinic/Renal Pathology Society consensus report on pathologic classification, diagnosis, and reporting of GNJ Am Soc Nephrol Sethi S Haas M Markowitz GS 12781287272016 https://doi.org/10.1681/ASN.20150606122656724310.1681/ASN.2015060612 PMC 4849835 · doi ↗ · pubmed ↗
- 5Renal outcome of Ig M nephropathy: a comparative prospective cohort study J Clin Med Chae Y Yoon HE Chang YK 4191102021 https://doi.org/10.3390/jcm 101841913457529810.3390/jcm 10184191 PMC 8466757 · doi ↗ · pubmed ↗
- 6Resolution of Ig M nephropathy after rituximab treatment Am J Kidney Dis Betjes MG Roodnat JI 10591062532009 https://doi.org/10.1053/j.ajkd.2008.10.0381908430910.1053/j.ajkd.2008.10.038 · doi ↗ · pubmed ↗
- 7Ig M nephropathy - successful treatment with rituximab Saudi J Kidney Dis Transpl Ahmed FA El-Meanawy A 235238302019 https://pubmed.ncbi.nlm.nih.gov/30804288/30804288 · pubmed ↗