The chromosomal genome sequence of the sponge Crambe crambe (Schmidt, 1862) and its associated microbial metagenome sequences
Manuel Maldonado, Lucia Pita, Ute Hentschel, Dirk Erpenbeck, Graeme Oatley, Elizabeth Sinclair, Eerik Aunin, Noah Gettle, Camilla Santos, Michael Paulini, Haoyu Niu, Victoria McKenna, Rebecca O’Brien, Paco Cardenas, Weizhi Song, Sihan Li, Shikai Liu

TL;DR
This paper presents the genome of the sponge Crambe crambe and the genomes of its associated microbes, providing insights into sponge biology and symbiosis.
Contribution
The study provides a high-quality chromosomal genome assembly of Crambe crambe and associated microbial metagenomes.
Findings
The host genome is 143.20 megabases long and scaffolded into 18 chromosomal pseudomolecules.
The mitochondrial genome is 19.53 kilobases in length.
Microbial genomes of symbiotic prokaryotes, including Beroebacter and Nitrosopumilus, were assembled.
Abstract
We present a genome assembly from an individual Crambe crambe (Porifera; Demospongiae; Poecilosclerida; Crambeidae). The host genome sequence is 143.20 megabases in span. Most of the assembly is scaffolded into 18 chromosomal pseudomolecules. The mitochondrial genome has also been assembled and is 19.53 kilobases in length. Several symbiotic prokaryotic genomes were assembled as MAGs, including two relevant sponge symbionts, the Candidatus Beroebacter blanensis/ AqS2 clade (Tethybacterales, Gammaproteobacteria) of LMA sponges, and the widely distributed archaeal Nitrosopumilus sp. clade.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1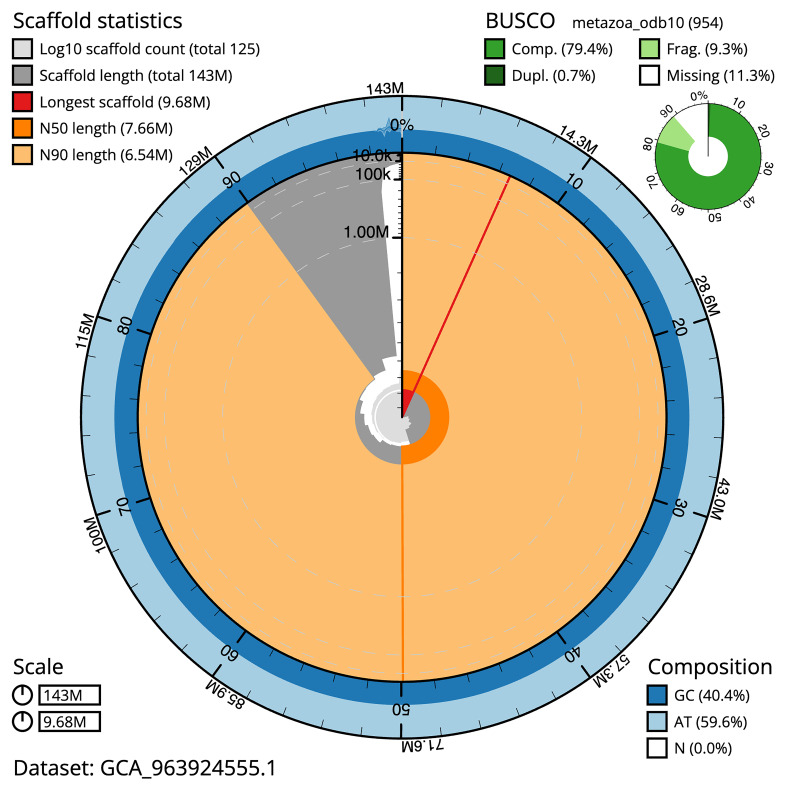 Figure 2
Figure 2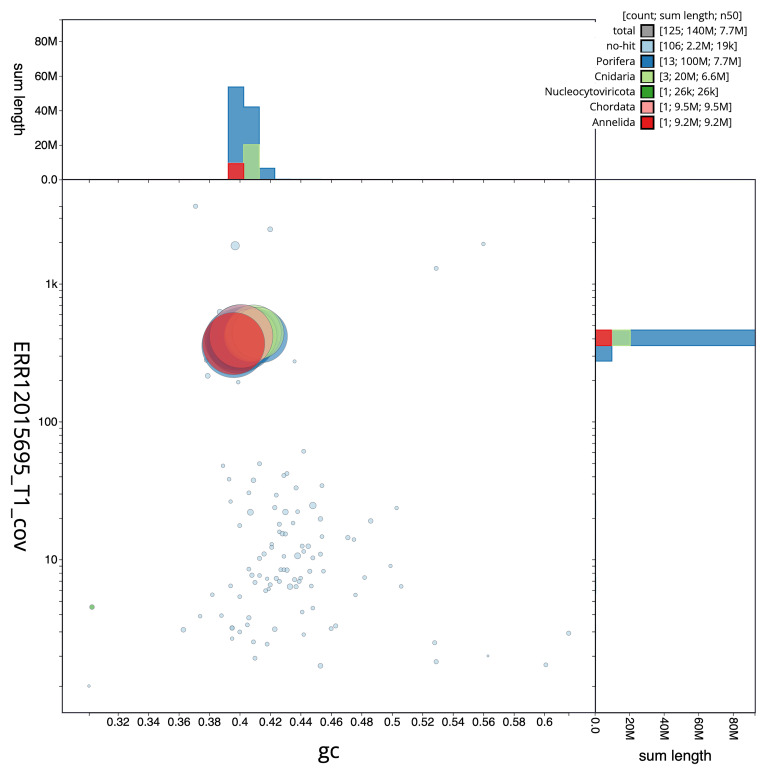 Figure 3
Figure 3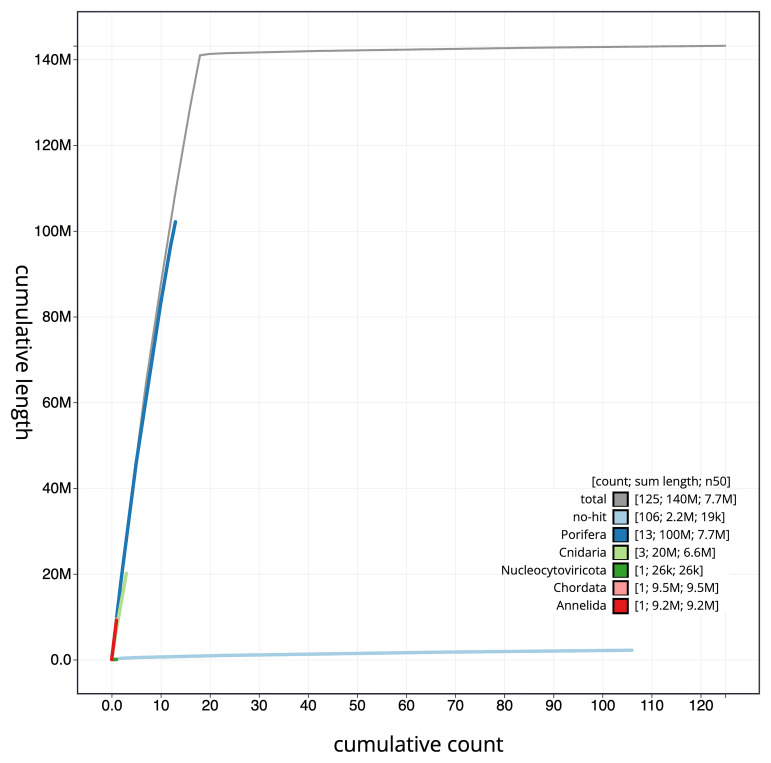 Figure 4
Figure 4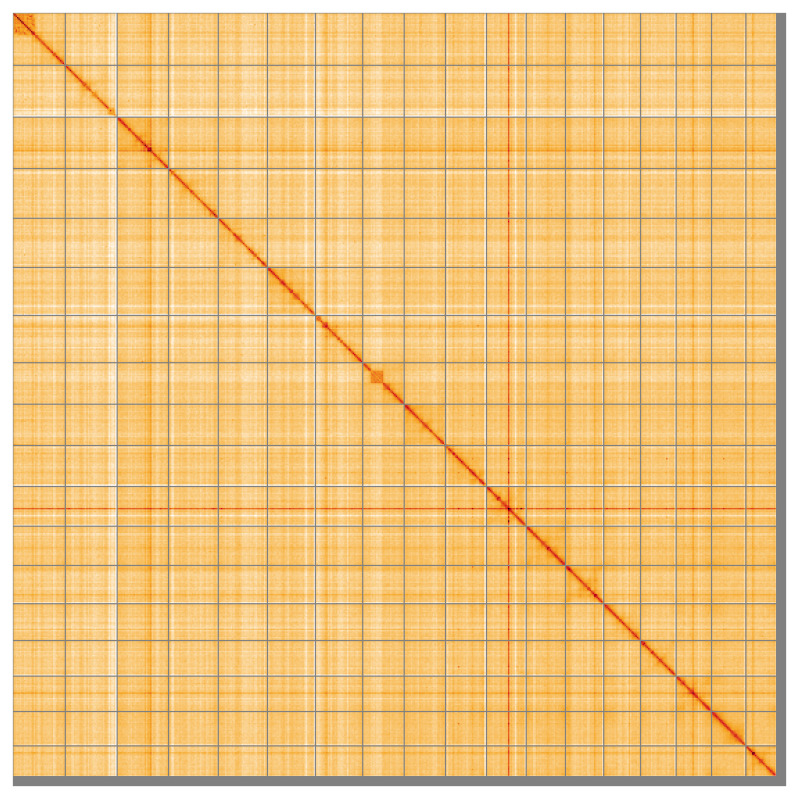 Figure 5
Figure 5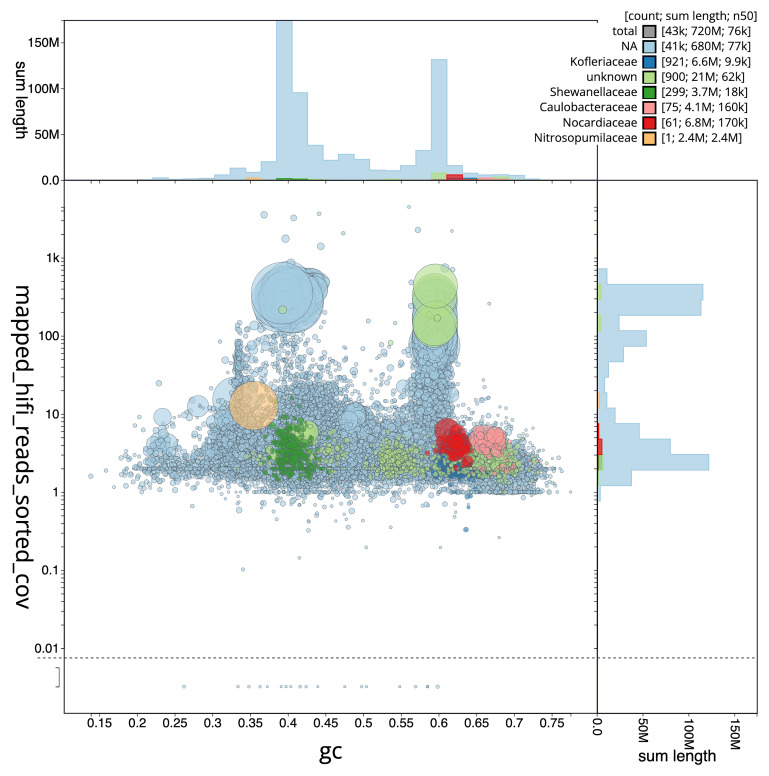 Figure 6
Figure 6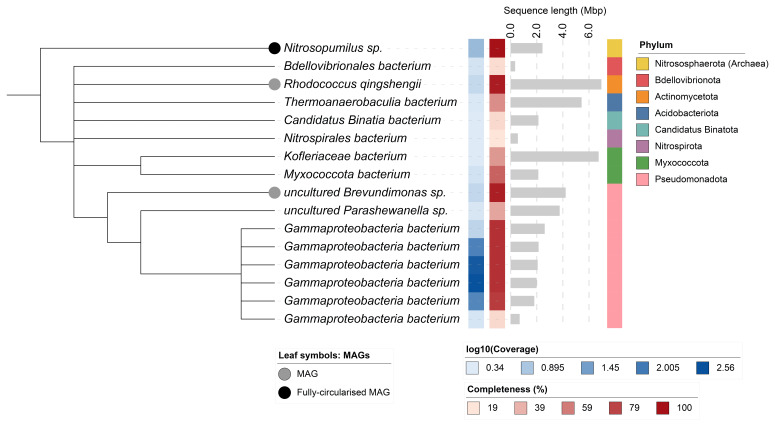 Figure 7
Figure 7| Project accession data | |||
|---|---|---|---|
| Assembly identifier | odCraCram1.1 | ||
| Species |
| ||
| Specimen | odCraCram1 | ||
| NCBI taxonomy ID | 3722 | ||
| BioProject | PRJEB65618 | ||
| BioSample ID | Genome sequencing: SAMEA9361910
| ||
| Isolate information | odCraCram1: (genome and Hi-C sequencing) | ||
| Assembly metrics | |||
| Consensus quality (QV) | 58.1 | ||
| BUSCO
| C:78.8%[S:78.0%,D:0.8%],F:9.4%,M:11.8%,n:954 | ||
| Percentage of assembly mapped
| 98.69% | ||
| Organelles | Mitochondrial genome: 19.53 kb | ||
| Sequencing information | |||
|
|
|
|
|
|
| ERR12512721 | 1.13e+09 | 170.77 |
|
| ERR12015695 | 9.82e+06 | 67.94 |
| Genome assembly | |||
| Assembly accession | GCA_963924555.1 | ||
|
| GCA_963924525.1 | ||
| Span (Mb) | 143.20 | ||
| Number of contigs | 178 | ||
| Contig N50 length (Mb) | 3.5 | ||
| Number of scaffolds | 124 | ||
| Scaffold N50 length (Mb) | 7.7 | ||
| Longest scaffold (Mb) | 9.77 | ||
| INSDC
| Name | Length
| GC% |
|---|---|---|---|
| 1 | 9.57 | 39.5 | |
| 2 | 9.68 | 40.0 | |
| 3 | 9.49 | 40.0 | |
| 4 | 9.08 | 40.0 | |
| 5 | 9.18 | 39.5 | |
| 6 | 8.84 | 40.0 | |
| 7 | 8.73 | 39.5 | |
| 8 | 7.66 | 40.0 | |
| 9 | 7.63 | 40.5 | |
| 10 | 7.57 | 40.5 | |
| 11 | 7.33 | 40.5 | |
| 12 | 7.24 | 41.0 | |
| 13 | 7.06 | 40.5 | |
| 14 | 6.8 | 40.5 | |
| 15 | 6.57 | 40.5 | |
| 16 | 6.54 | 41.5 | |
| 17 | 6.34 | 41.0 | |
| 18 | 5.67 | 40.5 | |
| MT | 0.02 | 37.0 |
| NCBI taxon | Taxid | GTDB taxonomy | Quality | Size (bp) | Contigs | Circular | Mean
| Completeness
| Contamination
|
|---|---|---|---|---|---|---|---|---|---|
| Nitrosopumilus sp. | 2024843 | g__Nitrosopumilus | High | 2,406,465 | 1 | Yes | 11.72 | 100.00 | 0.00 |
| uncultured
| 213418 | g__Brevundimonas | High | 4,185,465 | 75 | Partial | 4.39 | 95.06 | 4.27 |
| Rhodococcus qingshengii | 334542 | s__Rhodococcus
| High | 6,934,702 | 61 | No | 4.2 | 95.58 | 0.00 |
| Gammaproteobacteria
| 1913989 | f__AqS2 | Medium | 1,777,560 | 2 | No | 61.05 | 83.22 | 1.22 |
| Gammaproteobacteria
| 1913989 | f__AqS2 | Medium | 1,964,849 | 1 | Yes | 364.77 | 87.49 | 0.61 |
| Gammaproteobacteria
| 1913989 | f__AqS2 | Medium | 2,039,060 | 1 | Yes | 278.56 | 87.49 | 0.61 |
| Myxococcota bacterium | 2818507 | f__UBA6930 | Medium | 2,090,974 | 133 | No | 3.11 | 69.12 | 6.72 |
| Gammaproteobacteria
| 1913989 | f__AqS2 | Medium | 2,109,981 | 4 | No | 74.99 | 87.49 | 1.83 |
| Gammaproteobacteria
| 1913989 | g__UBA1858 | Medium | 2,576,596 | 26 | No | 4.79 | 87.52 | 1.97 |
| Thermoanaerobaculia
| 2651171 | f__UBA5704 | Medium | 5,408,624 | 319 | No | 2.54 | 52.97 | 1.44 |
| Bdellovibrionales
| 2053517 | g__JACOND01 | Low | 314,367 | 16 | No | 2.89 | 23.10 | 0.00 |
| Nitrospirales bacterium | 2358460 | g__Bin75 | Low | 524,459 | 58 | No | 2.22 | 19.47 | 0.00 |
| Gammaproteobacteria
| 1913989 | g__UBA1858 | Low | 655,358 | 63 | No | 2.67 | 24.99% | 1.77% |
| Candidatus Binatia
| 2838779 | g__JAAXHF01 | Low | 2,103,514 | 277 | No | 2.23 | 24.60% | 0.00% |
| uncultured
| 2547967 | g__Parashewanella | Low | 3,728,522 | 299 | Partial | 2.61 | 43.78% | 5.25% |
| Kofleriaceae bacterium | 2212474 | f__Haliangiaceae | Low | 6,724,497 | 921 | Partial | 2.17 | 48.48% | 5.11% |
| Software tool | Version | Source |
|---|---|---|
| BEDTools | 2.30.0 |
|
| bin3C | 0.3.3 |
|
| Blast | 2.14.0 |
|
| BlobToolKit | 4.3.7 |
|
| BUSCO | 5.4.3 and 5.5.0 |
|
| bwa-mem2 | 2.2.1 |
|
| CheckM | 1.2.1 |
|
| Cooler | 0.8.11 |
|
| DAS Tool | - |
|
| DIAMOND | 2.1.8 |
|
| dRep | 3.4.0 |
|
| fasta_windows | 0.2.4 |
|
| FastK | 427104ea91c78c3b8b8b49f1a7d6bbeaa869ba1c |
|
| GoaT CLI | 0.2.5 |
|
| GTDB-TK | 2.3.2 |
|
| Hifiasm | 0.19.5-r587 |
|
| HiGlass | 44086069ee7d4d3f6f3f0012569789ec138f42b84
|
|
| MaxBin | 2.7 |
|
| MerquryFK | d00d98157618f4e8d1a9190026b19b471055b22e |
|
| MetaBat2 | 2.15-15-gd6ea400 |
|
| MetaTOR | - |
|
| MitoHiFi | 2 |
|
| MultiQC | 1.14, 1.17, and 1.18 |
|
| Nextflow | 23.04.0-5857 |
|
| PretextView | 0.2 |
|
| PROKKA | 1.14.5 |
|
| purge_dups | 1.2.5 |
|
| samtools | 1.16.1, 1.17, and 1.18 |
|
| Seqtk | 1.3 |
|
| Singularity | 3.9.0 |
|
| TreeVal | 1.0.0 |
|
| YaHS | 1.1a.2 |
|
- —Wellcome Trust
- —Gordon and Betty Moore Foundation
- —Spanish Government
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMarine Sponges and Natural Products · Microbial Natural Products and Biosynthesis · Genomics and Phylogenetic Studies
Species taxonomy
Eukaryota; Opisthokonta; Metazoa; Porifera; Demospongiae; Heteroscleromorpha; Poecilosclerida; Crambeidae; Crambe (in: sponges) Crambe; Crambe crambe Vosmaer, 1880 (NCBI:txid3722).
Background
Crambe crambe ( Schmidt, 1862) is probably the most abundant sponge species in the sublittoral rocky bottoms of the Atlantic-Mediterranean region. It is a bright red encrusting sponge that grows at both well-lit and poorly lit sites, forming patches of up to 0.5 m ^2^ ( Pansini & Pronzato, 1990; Turon et al., 1998). As additional macroscopic clues for species identification, oscula and their radially converging excurrent channels are often visible on the sponge surface, which is slippery to the touch. The sponge grows not only on rocks, but also on barnacles and on the shells of the red oyster Spondylus gaederopus.
Due to its abundance, the species is ecologically important in many ways. For instance, its skeletal growth represents a substantial silicon sink for the sublittoral system ( Maldonado et al., 2005). The sponge also provides food and habitat for a variety of marine organisms, including recruitment habitat for juvenile ophiuroids ( Turon et al., 2000) and small benthic fish. C. crambe produces various bioactive compounds that interact chemically with many community members ( Becerro et al., 1994; Becerro et al., 1997), some of which have potential pharmaceutical applications derived from their antibacterial, antifungal, and anti-tumour properties, among others ( El-Demerdash et al., 2018). Given its biotechnological potential, attempts have been made to farm the species ( Padiglia et al., 2018). Despite its abundance and ecological versatility (or perhaps because of it), the species is thought to be a surviving relict of the Jurassic oceans. This hypothesis is supported by the observation that the formation of all four spicule types is only possible at a silicate concentration ≥100 µM – concentrations which are likely to have occurred in Jurassic seas before the ecological expansion of diatoms ( Maldonado et al., 1999). Secondly, the biogeographic distribution of the genus Crambe shows a clear Tethyan pattern ( Maldonado et al., 2001).
Regarding the microbiome, the sponge is a species with low microbial abundance. While most of the few microbes occur in low abundance extracellularly in the mesohyl and around the skeletal spongin fibres, some of the microbes have been documented by electron microscopy to be contained within vesicles in the cytoplasm of bacteriocytes that appear to contain a single microbial species per cell ( Carrier et al., 2022; Maldonado, 2007). Gammaproteobacteria, ammonia-oxidising Nitrosopumilus sp. (Archaea) and a single taxon, Candidatus Beroebacter blanensis, dominate the microbial community . This latter symbiont clade appears to be vertically transmitted ( Turon et al., 2024). It was originally classified as Betaproteobacteria ( Croué et al., 2013), but was later identified as Ca. Beroebacter blanensis, belonging to a novel bacterial order, Candidatus ( Ca.) Tethybacterales within the Gammaproteobacteria and consisting mainly of sponge symbionts ( Taylor et al., 2021). The well characterized symbiont “AqS2” of Amphimedon queenslandica is the nearest phylogenetic relative of the B. blanensis clade, which displays genome reduction and limited metabolic capabilities, likely reflecting an adaptation to a symbiotic lifestyle within the sponge host ( Gauthier et al., 2016).
The sexual condition of the species is hermaphroditism. It is worth noting that its spermatozoa are highly atypical within the phylum. They are very elongated and V-shaped, with the flagellum inserted in an antero-lateral position next to a true acrosome ( Riesgo & Maldonado, 2009; Tripepi et al., 1984). This general organisation of the spermatozoon, which closely resembles that of Phoronida spermatozoa, appears to be common in the order Poecilosclerida but not in other sponges. Fertilisation is internal, and embryos are incubated for several months, until they develop into bright red, non-tufted parenchymella larvae ( Maldonado & Bergquist, 2002; Uriz et al., 2001). In western Mediterranean populations, larval release extends from mid-July to mid-August, and larval production can be as high as 76 embryos per cm ^2 ^of sponge tissue ( Uriz et al., 1998), which would explain the abundance of adults.
The sequencing of the whole-chromosome genome of C. crambe will facilitate in-depth understanding of the genomic basis of this species biology, as well as its ecology and evolution. This genome will be particularly useful for investigating the evolution of sexual strategies in Demospongiae, as well as for clarifying between-family relationships within the order Poecilosclerida. Together with the genome sequences of C. crambe microbial symbionts presented here, the novel data will enable targeted examination of the molecular basis of sponge silicate metabolism and skeleton formation, alkaloid metabolism, and sponge-microbe interactions in the role of carbon cycling, among other key questions in sponge symbiosis.
Genome sequence report
The genome was sequenced from an adult Crambe crambe ( Figure 1) collected from Blanes, Girona, Spain. A total of 459-fold coverage in Pacific Biosciences single-molecule HiFi long reads was generated. Primary assembly contigs were scaffolded with chromosome conformation Hi-C data. Manual assembly curation corrected 62 missing joins or mis-joins and removed 18 haplotypic duplications, reducing the assembly length by 2.19% and the scaffold number by 29.78%, also decreasing the scaffold N50 by 0.31%.
Photograph of the Crambe crambe (odCraCram1) specimen used for genome sequencing.
The final assembly has a total length of 143.20 Mb in 124 sequence scaffolds with a scaffold N50 of 7.7 Mb ( Table 1). The snail plot in Figure 2 provides a summary of the assembly statistics, while the distribution of assembly scaffolds on GC proportion and coverage is shown in Figure 3. The cumulative assembly plot in Figure 4 shows curves for subsets of scaffolds assigned to different phyla. Most (98.69%) of the assembly sequence was assigned to 18 chromosomal-level scaffolds. Chromosome-scale scaffolds confirmed by the Hi-C data are named in order of size ( Figure 5; Table 2). While not fully phased, the assembly deposited is of one haplotype. Contigs corresponding to the second haplotype have also been deposited. The mitochondrial genome was also assembled and can be found as a contig within the multifasta file of the genome submission.
Table 1.: Genome data for Crambe crambe, odCraCram1.1.
Genome assembly of Crambe crambe, odCraCram1.1: metrics.The BlobToolKit Snailplot shows N50 metrics and BUSCO gene completeness. The main plot is divided into 1,000 size-ordered bins around the circumference with each bin representing 0.1% of the 143,197,480 bp assembly. The distribution of scaffold lengths is shown in dark grey with the plot radius scaled to the longest scaffold present in the assembly (9,683,886 bp, shown in red). Orange and pale-orange arcs show the N50 and N90 scaffold lengths (7,656,483 and 6,535,638 bp), respectively. The pale grey spiral shows the cumulative scaffold count on a log scale with white scale lines showing successive orders of magnitude. The blue and pale-blue area around the outside of the plot shows the distribution of GC, AT and N percentages in the same bins as the inner plot. A summary of complete, fragmented, duplicated and missing BUSCO genes in the metazoa_odb10 set is shown in the top right. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/Crambe_crambe/dataset/GCA_963924555.1/snail.
Genome assembly of Crambe crambe, odCraCram1.1: BlobToolKit GC-coverage plot.Scaffolds are coloured by phylum. Circles are sized in proportion to scaffold length. Histograms show the distribution of scaffold length sum along each axis. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/Crambe_crambe/dataset/GCA_963924555.1/blob.
Genome assembly of Crambe crambe, odCraCram1.1: BlobToolKit cumulative sequence plot.The grey line shows cumulative length for all scaffolds. Coloured lines show cumulative lengths of scaffolds assigned to each phylum using the buscogenes taxrule. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/Crambe_crambe/dataset/GCA_963924555.1/cumulative.
Genome assembly of Crambe crambe, odCraCram1.1: Hi-C contact map of the odCraCram1.1 assembly, visualised using HiGlass.Chromosomes are shown in order of size from left to right and top to bottom. An interactive version of this figure may be viewed at https://genome-note-higlass.tol.sanger.ac.uk/l/?d=IeGb4iyXTOqWeiUVkpdMrA.
Table 2.: Chromosomal pseudomolecules in the genome assembly of Crambe crambe, odCraCram1.
The estimated Quality Value (QV) of the final assembly is 58.1. The assembly has a BUSCO v5.4.3 completeness of 78.8% (single = 78.0%, duplicated = 0.8%), using the metazoa_odb10 reference set ( n = 954).
Metagenome report
Sixteen binned genomes were generated from the metagenome assembly ( Figure 6), of which three were classified as high-quality metagenome assembled genomes (MAGs) (see methods). The completeness values for these assemblies range from approximately 20% to 100% with contamination below 7%. A cladogram of the binned metagenomes is shown in Figure 7. For details on binned genomes see Table 3.
Blob plot of base coverage in mapped against GC proportion for sequences in the metagenome of Crambe crambe.Binned metagenomes are coloured by family. Circles are sized in proportion to sequence length on a square root scale, ranging from 501 to 4,126,685. Histograms show the distribution of sequence length sum along each axis An interactive version of this figure may be viewed here.
Cladogram showing the taxonomic placement of metagenome bins, constructed using NCBI taxonomic identifiers with taxonomizr and annotated in iTOL.Colours indicate phylum-level taxonomy. Additional tracks show sequencing coverage (log₁₀), estimated genome size (Mbp), and completeness. Bins that meet the criteria for MAGs are marked with a grey circle; the single fully circularised MAG is marked in black.
Methods
Sample acquisition
A specimen of Crambe crambe (specimen ID GHC0000181, ToLID odCraCram1) was collected from Blanes, Girona, Spain (latitude 41.67, longitude 2.80) on 2021-02-01 by SCUBA diving. The specimen was collected and identified by Manuel Maldonado (CEAB-CSIC) and preserved by snap-freezing.
Nucleic acid extraction
The workflow for high molecular weight (HMW) DNA extraction at the Wellcome Sanger Institute (WSI) Tree of Life Core Laboratory includes a sequence of core procedures: sample preparation; sample homogenisation, DNA extraction, fragmentation, and clean-up. Protocols are available on protocols.io ( Denton et al., 2023). In sample preparation, the odCraCram1 sample was weighed and dissected on dry ice ( Jay et al., 2023). Prior to DNA extraction, the sponge sample was bathed in “L buffer” (10 mM Tris, pH 7.6, 100 mM EDTA, 20 mM NaCl), minced into small pieces using a scalpel and the cellular interior separated from the mesohyl using forceps ( Lopez, 2022). HMW DNA was extracted using the Manual MagAttract v1 protocol ( Strickland et al., 2023b). DNA was sheared into an average fragment size of 12–20 kb in a Megaruptor 3 system ( Todorovic et al., 2023). Sheared DNA was purified by solid-phase reversible immobilisation ( Strickland et al., 2023a), using AMPure PB beads to eliminate shorter fragments and concentrate the DNA. The concentration of the sheared and purified DNA was assessed using a Nanodrop spectrophotometer, Qubit Fluorometer and Qubit dsDNA High Sensitivity Assay kit. Fragment size distribution was evaluated by running the sample on the FemtoPulse system.
Sequencing
Pacific Biosciences HiFi circular consensus DNA sequencing libraries were constructed according to the manufacturers’ instructions. DNA sequencing was performed by the Scientific Operations core at the WSI on a Pacific Biosciences Revio instrument. Hi-C data were also generated from tissue of odCraCram1 using the Arima2 kit and sequenced on the Illumina NovaSeq 6000 instrument.
Host genome assembly and curation
Assembly was carried out with Hifiasm ( Cheng et al., 2021) and haplotypic duplication was identified and removed with purge_dups ( Guan et al., 2020). The assembly was then scaffolded with Hi-C data ( Rao et al., 2014) using YaHS ( Zhou et al., 2023). The mitochondrial genome was assembled using MitoHiFi ( Uliano-Silva et al., 2023), which runs MitoFinder ( Allio et al., 2020) and uses these annotations to select the final mitochondrial contig and to ensure the general quality of the sequence. Table 4 contains a list of relevant software tool versions and sources.
The assembly was checked for contamination and corrected using the TreeVal pipeline ( Pointon et al., 2023). Manual curation was primarily conducted using PretextView ( Harry, 2022), with additional insights provided by JBrowse2 ( Diesh et al., 2023) and HiGlass ( Kerpedjiev et al., 2018). Any identified contamination, missed joins, and mis-joins were corrected, and duplicate sequences were tagged and removed. The curation process is documented at https://gitlab.com/wtsi-grit/rapid-curation.
Taxonomic verification
Molecular markers obtained from the assembly were used to reconstruct the phylogenetic position of the sample. In an alignment using MAFFT v7.450 ( Katoh & Standley, 2013), the COI barcoding fragment (“Folmer” fragment) of the sample was found to be identical to haplotype 1 from a dedicated study on Crambe crambe ( Duran et al., 2004, AF526297), besides samples from other studies on this species as published in NCBI Genbank.
Host assembly quality assessment
The Merqury.FK tool ( Rhie et al., 2020), run in a Singularity container ( Kurtzer et al., 2017), was used to evaluate k-mer completeness and assembly quality for the primary and alternate haplotypes using the k-mer databases ( k = 31) that were computed prior to genome assembly. The analysis outputs included assembly QV scores and completeness statistics.
A Hi-C contact map was produced for the final version of the assembly. The Hi-C reads were aligned using bwa-mem2 ( Vasimuddin et al., 2019) and the alignment files were combined using SAMtools ( Danecek et al., 2021). The Hi-C alignments were converted into a contact map using BEDTools ( Quinlan & Hall, 2010) and the Cooler tool suite ( Abdennur & Mirny, 2020). The contact map is visualised in HiGlass ( Kerpedjiev et al., 2018).
The blobtoolkit pipeline is a Nextflow port of the previous Snakemake Blobtoolkit pipeline ( Challis et al., 2020). It aligns the PacBio reads in SAMtools and minimap2 ( Li, 2018) and generates coverage tracks for regions of fixed size. In parallel, it queries the GoaT database ( Challis et al., 2023) to identify all matching BUSCO lineages to run BUSCO ( Manni et al., 2021). For the three domain-level BUSCO lineages, the pipeline aligns the BUSCO genes to the UniProt Reference Proteomes database ( Bateman et al., 2023) with DIAMOND blastp ( Buchfink et al., 2021). The genome is also divided into chunks according to the density of the BUSCO genes from the closest taxonomic lineage, and each chunk is aligned to the UniProt Reference Proteomes database using DIAMOND blastx. Genome sequences without a hit are chunked using seqtk and aligned to the NT database with blastn ( Altschul et al., 1990). The blobtools suite combines all these outputs into a blobdir for visualisation.
The blobtoolkit pipeline was developed using nf-core tooling ( Ewels et al., 2020) and MultiQC ( Ewels et al., 2016), relying on the Conda package manager, the Bioconda initiative ( Grüning et al., 2018), the Biocontainers infrastructure ( da Veiga Leprevost et al., 2017), as well as the Docker ( Merkel, 2014) and Singularity ( Kurtzer et al., 2017) containerisation solutions.
Metagenome assembly
The metagenome assembly was generated using metaMDBG ( Benoit et al., 2024) and binned using MetaBAT2 ( Kang et al., 2019), MaxBin ( Wu et al., 2014), bin3C ( DeMaere & Darling, 2019), and MetaTOR. The resulting bin sets of each binning algorithm were optimised and refined using DAS Tool ( Sieber et al., 2018). PROKKA ( Seemann, 2014) was used to identify tRNAs and rRNAs in each bin, CheckM ( Parks et al., 2015) (checkM_DB release 2015-01-16) was used to assess bin completeness/contamination, and GTDB-TK ( Chaumeil et al., 2022) (GTDB release 214) was used to taxonomically classify bins. Taxonomic replicate bins were identified using dRep ( Olm et al., 2017), with default settings (95% ANI threshold). The final bin set was filtered for bacteria and archaea. All bins were assessed for quality and categorised as metagenome-assembled genomes (MAGs) if they met the following criteria: contamination ≤ 5%, presence of 5S, 16S, and 23S rRNA genes, at least 18 unique tRNAs, and either ≥ 90% completeness or ≥ 50% completeness with fully circularised chromosomes. Bins that did not meet these thresholds, or were identified as taxonomic replicates of MAGs, were retained as ‘binned metagenomes’ provided they had ≥ 50% completeness and ≤ 10% contamination. A cladogram based on NCBI taxonomic assignments was generated using the ‘taxonomizr’ package in R. The tree was visualised and annotated using iTOL ( Letunic & Bork, 2024). Software tool versions and sources are given in Table 4.
Wellcome Sanger Institute – Legal and Governance
The materials that have contributed to this genome note have been supplied by a Tree of Life collaborator. The Wellcome Sanger Institute employs a process whereby due diligence is carried out proportionate to the nature of the materials themselves, and the circumstances under which they have been/are to be collected and provided for use. The purpose of this is to address and mitigate any potential legal and/or ethical implications of receipt and use of the materials as part of the research project, and to ensure that in doing so we align with best practice wherever possible. The overarching areas of consideration are:
• Ethical review of provenance and sourcing of the material
• Legality of collection, transfer and use (national and international)
Each transfer of samples is undertaken according to a Research Collaboration Agreement or Material Transfer Agreement entered into by the Tree of Life collaborator, Genome Research Limited (operating as the Wellcome Sanger Institute) and in some circumstances other Tree of Life collaborators.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abdennur N Mirny LA : Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 2020;36(1):311–316. 10.1093/bioinformatics/btz 540 31290943 PMC 8205516 · doi ↗ · pubmed ↗
- 2Allio R Schomaker-Bastos A Romiguier J : Mito Finder: efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol Ecol Resour. 2020;20(4):892–905. 10.1111/1755-0998.13160 32243090 PMC 7497042 · doi ↗ · pubmed ↗
- 3Altschul SF Gish W Miller W : Basic Local Alignment Search Tool. J Mol Biol. 1990;215(3):403–410. 10.1016/S 0022-2836(05)80360-2 2231712 · doi ↗ · pubmed ↗
- 4Bateman A Martin MJ Orchard S : Uni Prot: the universal protein knowledgebase in 2023. Nucleic Acids Res. 2023;51(D 1):D 523–D 531. 10.1093/nar/gkac 1052 36408920 PMC 9825514 · doi ↗ · pubmed ↗
- 5Becerro MA Uriz MJ Turón X : Trends in space occupation by the encrusting sponge Crambe crambe: variation in shape as a function of size and environment. Mar Biol. 1994;121:301–307. 10.1007/BF 00346738 · doi ↗
- 6Becerro MA Uriz MJ Turon X : Chemically-mediated interactions in benthic organisms: the chemical ecology of Crambe crambe (Porifera, Poecilosclerida). Hydrobiologia. 1997;355(1–3):77–89. 10.1023/A:1003019221354 · doi ↗
- 7Benoit G Raguideau S James R : High-quality metagenome assembly from long accurate reads with meta MDBG. Nat Biotechnol. 2024;42(9):1378–1383. 10.1038/s 41587-023-01983-6 38168989 PMC 11392814 · doi ↗ · pubmed ↗
- 8Buchfink B Reuter K Drost HG : Sensitive protein alignments at Tree-of-Life scale using DIAMOND. Nat Methods. 2021;18(4):366–368. 10.1038/s 41592-021-01101-x 33828273 PMC 8026399 · doi ↗ · pubmed ↗